

75 группа 2 вариант / Химия / fischem
.pdfВ зависимости от числа молекул, участвующих в элементарном акте,
реакции называются мономолекулярными, бимолекулярными, тримолекулярными. Число частиц указывает молекулярность реакции.
Если в реакцию вступает более трех молекул, то значит, что она идет через ряд последовательных стадий, в каждой из которых участвует 1-2 молекулы. Для сложных реакций понятие молекулярности не имеет смысла.
|
|
|
Таблица 5.1. |
|
Примеры реакций различной молекулярности |
|
|||
Мономолекулярные |
Бимолекулярные |
Тримолекулярные реакции |
||
реакции |
реакции |
|
|
|
А→В+ другие продукты |
A+B→D+другие |
2NO+O2=2NO2 |
|
|
(СН2)3 →СН3-СН=СН2 |
продукты |
I+I+Ar=I2+Ar-148Дж/мольК, |
||
300о |
|
|
|
|
циклопропан пропилен |
NO2+CO=NO+CO2 |
где |
аргон |
отводит |
Br2→2Br |
NO+O3=NO2+O2 |
выделяющуюся энергию. |
||
Кинетика простых реакций.
Когда уравнение реакции точно отражает ее ход, основной закон химической кинетики – закон действия масс формулируется так: скорость реакции прямо пропорциональна произведению концентрации реагирующих веществ в степени их стехиометрических коэффициентов.
V=-d[A]/dτ=k[A]a[B]b, |
(5.4) |
где a,b – частные кинетические порядки, a+b=n - общий кинетический |
|
порядок реакции. |
|
Частные кинетические порядки |
реакции в общем случае не равны |
стехиометрическим коэффициентам, они определяются экспериментально. В отличие от молекулярности порядки реакции могут принимать дробные, нулевые, отрицательные значения. Так скорость разложения уксусного альдегида в газовой фазе пропорциональна его концентрации в степени 3/2:
СН3СНO → СН4+СО; -d[CH3CHO]/dτ=k[CH3CHO]3/2, откуда n=3/2.
Если |
реакции |
протекают по нулевому порядку, |
то для нее: |
||
v = − |
dC |
= kC 0 |
= k . |
|
(5.5) |
dτ |
|
||||
|
|
|
|
|
|
Перепишем выражение в виде: |
|
||||
dC = −kdτ , |
|
|
|
||
возьмем неопределенный интеграл от обеих частей уравнения |
|
||||
∫dC = −∫kdτ , |
|
|
|
||
тогда |
|
|
|
|
|
C = −kτ + B , |
где |
В – постоянная интегрирования, |
концентрация в |
||
начальный момент времени С0=В. Тогда получаем: |
|
||||
C = C0 − kτ . |
|
|
(5.6) |
||
Следовательно, в реакциях нулевого порядка концентрация линейно уменьшается со временем.
Константа скорости реакции нулевого порядка вычисляется по формуле:
k = |
|
1 |
(C0 −C) , |
(5.7) |
|
τ |
|||||
|
|
|
|||
91
где С0 – начальная концентрация, С – концентрация в момент времени τ. Константа скорости измеряется в [моль/л c].
Нулевой и отрицательный порядки встречаются в гетерогеннокаталитических реакциях. Кажущийся нулевой порядок может быть обусловлен большим избытком исходного вещества. Так для инверсии сахарозы скорость реакции выражается уравнением: v=k[C12H22O11], т.к. концентрация воды в 0,1 М растворе остается в ходе реакции практически неизменной, и во много раз превышает количество воды, участвующее в реакции (0,1 моль/л).
Поэтому часто экспериментальное определение частного кинетического порядка реакции по какому-то из веществ, проводят в условиях постоянной концентрации другого вещества: [B]≥20[A], v=k1[A]a, где k1=k[B]b. Проводя реакцию при двух различных концентрациях вещества А, находят скорости v1 и
v2:
v1=k1[A1]n, v2=k1[A2] n, v2/v1=([A2]/[A1])n,
откуда
|
lg(v2 / v1 ) |
|
||
n = |
|
. |
(5.8) |
|
lg([A ]/[A ]) |
||||
2 |
1 |
|
|
|
Кинетическое уравнение реакции первого порядка имеет вид:
v = − |
dC |
= kC , |
(5.9) |
||||
|
|||||||
|
|
|
dτ |
|
|
|
|
Отделим переменные и проинтегрируем: |
|||||||
|
dC |
= −kdτ, |
∫ |
dC |
= −∫kdτ , ln C = −kτ + B . |
||
|
|
C |
|||||
|
C |
|
|
||||
Находим постоянную интегрирования В, считая, что в начальный момент времени концентрация реагента С=С0, ln C0 = B , поэтому
ln C = ln C0 − kτ . |
(5.10) |
Следовательно, для реакций первого порядка характерна линейная зависимость логарифма концентрации от времени.
Константа скорости реакции первого порядка равна:
k |
= |
|
1 |
ln |
C0 |
|
или k = 2,303 |
1 |
lg |
C0 |
|
(5.11) |
||||||||
τ |
C |
τ |
C |
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
и измеряется [с-1]. |
|
|
|
|
|
|||||||||||||||
Кинетическое уравнение реакций второго порядка: |
|
|||||||||||||||||||
1 |
= |
|
1 |
|
|
|
+ kτ . |
|
|
|
|
(5.12) |
||||||||
|
C |
C0 |
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
Константа скорости вычисляется по формуле: |
|
|||||||||||||||||||
|
|
|
1 |
|
1 |
|
|
1 |
|
|
|
|
|
(5.13) |
||||||
k |
= |
|
|
− |
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
τ |
C |
|
|
C0 |
|
|
|
|
|
||||||||
измеряется в [л/моль с].
Порядки реакции могут быть вычислены способом подстановки. Экспериментально определяют концентрации исходных веществ в фиксируемые моменты времени и подставляют последовательно в уравнения констант скорости нулевого, первого, второго или третьего порядков. То уравнение, которое дает близкие значения константы скорости, определяет
92

порядок реакции. Можно графическим методом в координатах С - τ, lnС - τ, 1/С-τ, 1/С2-τ. Порядок изучаемой реакции будет соответствовать тому уравнению, для которого точки на графике ложатся на прямую линию.
Рис.13. Изменение концентрации со временем в реакциях: а – нулевого порядка, б – первого порядка, в – второго порядка, г - третьего порядка.
Порядки реакции могут быть вычислены также графически в логарифмических координатах:
lg v=lg k+n lgС (5.14)
На графике log v – log С порядок реакции равен тангенсу наклона получаемой прямой.
Зависимость скорости химической реакции от температуры.
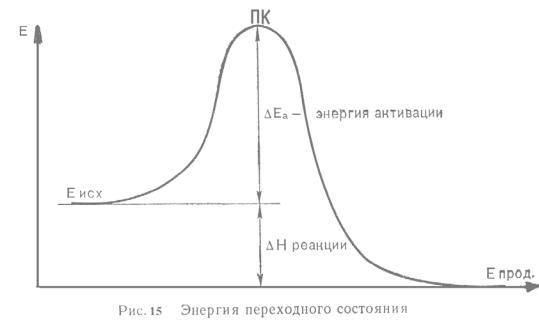
Скорость большинства реакций увеличивается с температурой. Увеличение скорости реакции с температурой описывается уравнением Аррениуса:
k=Aexp(-E/RT), (5.15)
где k-константа скорости реакции, А – предэкспоненциальный множитель, Е – энергия активации.
Рис.14. Линейная зависимость log K разложения HI от 1/Т
Если значения констант скорости реакции определены для нескольких температур, то график зависимости lgk от T-1 по уравнению:
ln k=ln A-(E/R)T-1
или
lg k=lg A-(E/2,303R)T-1 |
(5.16) |
93
имеет вид прямой линии. Она отсекает на оси ординат отрезок равный lg A, из тангенса наклона прямой определяется величину Е в кДж/моль:
E = 2,303R |
lg k1 −lg k2 |
. |
(5.17) |
|
|
||||
|
(T −1 |
−T −1 ) |
|
|
1 |
2 |
|
|
|
Для различных реакций энергия активации изменяется в пределах 40 – 300 кДж/моль. При этом повышение температуры реакции на 10 градусов ведет к увеличению скорости в 2-4 раза. Что в соответствии с гипотезой Аррениуса, объясняется существованием активных молекул, обладающих энергией, значительно превосходящей среднюю энергию молекул, а потому более реакционноспособных.
Множитель ехр(-E/RT) – доля молекул, обладающих энергией, равной или большей энергии активации.
Согласно теории переходного состояния (активированного комплекса) условием реакции является образование непрочного промежуточного комплекса между исходными веществами:
А2+В2 Z,
где Z- активный промежуточный комплекс. Равновесие этого процесса характеризуется константой:
K=[Z]/[A2][B2]. |
(5.18) |
Константа связана со свободной энергией Гиббса: |
|
G0=-RTlnK |
(5.19) |
или |
|
K=e- G/RT. |
(5.20) |
Т.к. G= H-T S, заменив в уравнении |
G, получим: |
K=e S/Re- H/RT. |
(5.21) |
Тогда |
|
[Z]= e S/Re- H/RT[A2][B2]. |
(5.22) |
94
Скорость распада промежуточного комплекса на продукты реакции АВ можно выразить как скорость бимолекулярной реакции и как скорость
мономолекулярной: |
|
v=K2[A2][B2]=K3[Z], |
(5.23) |
где К3 – константа скорости распада переходного комплекса. Из |
|
предыдущих уравнений получаем |
|
К2=К3 e S/Re- H/RT. |
(5.24) |
Полагают, что константа скорости распада активированного комплекса равна частоте колебаний разрывающейся связи промежуточного комплекса.
К3=kT/h, |
(5.25) |
где k- константа Больцмана, h – постоянная Планка. Подставим в |
|
предыдущее уравнение: |
|
К2=kT/h e S/Re- H/RT. |
(5.26) |
Прологарифмируем и получим: l |
|
lnK2=lnkT/h + S/R - Н/RТ, |
(5.27) |
сравним с уравнением Аррениуса: lnK2=lnA-E/RT.
Следовательно А=kT/h e S/R, Е= Н, т.е. энергия активации равна энтальпии образования промежуточного комплекса. Следовательно, величина предэкспоненциального множителя зависит от энтропии образования промежуточного комплекса, а т.к. S<0, то e S/R<1. Доля результативных столкновений молекул всегда меньше общего числа столкновений.
Цепные реакции.
Примером цепных реакций являются реакции взаимодействия хлора с водородом, реакции хлорирования предельных углеводородов, окисление ненасыщенных углеводородов нефти, прогоркание масла и др. Смесь водорода и хлора взрывается при облучении светом или при введении в нее кусочка натрия. Свет и натрий возбуждают цепную реакцию – последовательность большого числа элементарных реакций, в каждой из которых заново образуется свободный атом, радикал, т.е. частица с большой реакционной способностью, которая не требуют активации.
Na + Cl2 → Cl• + NaCl
Cl2 + hν → Cl• + Cl•
Cl• + H2 → HCl + H•
H• + Cl2→ HCl + Cl• Cl• + H• → HCl
H• + H• → H2
Cl• + Cl•→ Cl2
На место прореагировавшего радикала образуется один свободный радикал. Новые радикалы могут образовываться только в результате инициирования. Такой механизм реакции называется – неразветвленной реакцией.
Разветвленная реакция характеризуется увеличением числа свободных радикалов независимо от процесса инициирования. Примером такой реакции является одна из элементарных стадий горения водорода в кислороде:
95
О•+Н2 →НО• + Н•.
Горение бензина в автомобильном двигателе представляет собой высокотемпературный крекинга углеводородов до радикалов и взаимодействие этих радикалов с молекулярным кислородом, который представляет собой бирадикал.
Цепные реакции исследованы академиком Семеновым Н.Н.
Цепная реакция характеризуется длиной цепи – числом звеньев следующих за инициацией. Для реакции водорода с хлором на каждый поглощенный квант света образуется 106 молекул HCl (Беденштейн, 1913). Эту реакцию можно отнести к фотохимическим.
Фотохимические реакции
Реакции, которые возникают или ускоряются под действием света. К этим реакциям относятся реакции фотосинтеза в хлоропластах зеленого листа. Все виды органических топлив на Земле возникли благодаря фотохимическим реакциям.
Энергия света должна быть поглощена молекулой реагирующего соединения (закон Гроттгуса – Дрейпера). Открытие квантовой природы света привела И.Штарка и А.Энштейна к формулировке второго закона фотохимии: превращение одной молекулы требует поглощения одного кванта света. Квантовым выходом (γ) называют отношение числа молекул, прореагировавших в фотохимической реакции, к числу поглощенных квантов. По закону Штарка – Эйнштейна квантовый выход всегда равен 1. Но это относится только к первичному процессу поглощения света – перехода молекул в возбужденное состояние. Поскольку молекула может дезактивироваться в результате испускания или передачи поглощенной энергии при столкновении с другой молекулой, γ может быть меньше 1. В цепных реакциях γ>1.
Фотохимические реакции могут протекать по двум механизмам: 1)механизм возбуждения – время жизни активированной молекулы
достаточно велико, она может реагировать с другими молекулами, может дезактивироваться, испустив квант света.
а)А+hν→A* (возбуждение);
б) A*→ А+hν (флуоресценция) ;
в) A* +А→А2 (димеризация).
2)диссоциативный механизм – время жизни возбужденной молекулы не превышает времени одного внутримолекулярного колебания, возбужденная молекула практически сразу диссоциирует на две части:
HI+hν→H+I;
H+HI→H2+I;
I+I→I2.
Т.к. две последние стадии идут без поглощения света, и в них участвует еще одна молекула HI, то γ=2.
Фотосенсибилизация – поглощение света сенсибилизаторами
(световыми катализаторами) и передача его другим молекулам, претерпевающим в результате химические превращения.
96
Hg+hν→Hg•; Hg•+H2→Hg+H+H.
Катализ
Катализ – ускорение химической реакции в присутствии катализаторов – веществ, которые в реакции не расходуются и не входят в состав продуктов.
Предложил этот термин И.Я. Берцелиус (1779-1848).
Различают: гомогенный катализ – реагирующие вещества и катализатор находятся в одной фазе; гетерогенный катализ – катализатор находится в виде твердого вещества, реакция идет на поверхности раздела твердой фазы и жидкой или газообразной.
Катализаторы не влияют на термодинамику реакции, т.е. не изменяют энтальпию и энергию Гиббса реакции. Если энергия Гиббса реакции положительна, то в присутствии катализаторов эта реакция не станет самопроизвольной. Катализаторы могут ускорять наступление химического равновесия, но не влияют на константу равновесия. Катализатор увеличивает константу скорости химической реакции. Так как константа равновесия равна отношению констант скоростей прямой и обратной реакций, и от катализатора не зависит, следовательно, катализатор в одинаковой степени влияет на константу скоростей прямой и обратной реакций.
Катализаторы обладают селективностью – способностью ускорять одну из нескольких возможных реакций.
nCO+2(n+1)H2 |
Co+ThO2 +MgO |
|
CnH2n+2+nH2O |
|
→ |
|
|
CO+3H2 |
Ni |
|
CH4+H2O |
→ |
|
||
CO+2H2 |
ZnO+Cr2O3 |
|
CH3OH |
|
→ |
|
|
Действие катализатора основано |
на образовании промежуточного |
||
комплекса с реагирующим веществом. |
Энергия образования промежуточного |
||
комплекса много ниже энергии активации некаталитической реакции: |
|||
А + К → АК |
|
|
|
АК + В → АВ + К.
1)Примером гомогенного катализа является процесс, протекающий в производстве серной кислоты башенным способом. Этот процесс идет в газовой фазе:
SO2 + NO2→SO3 + NO, NO + 1/2O2 → NO2.
2)Кислотно-основной катализ в растворах. Роль кислот и оснований состоит в образовании с реагентом промежуточных соединений за счет донорно-акцепторного взаимодействия. При этом образуются центры с повышенной или пониженной электронной плотностью. Механизм кислотноосновного катализа всегда включает стадию протонирования реагирующей молекулы кислотой, и стадию депротонирования, обусловленную взаимодействием с основанием. В соответствии с теорией Бернстеда кислотой
97
является частица, которая отдает протон, а основанием – частица, принимающая протон. При этом кислота становится основанием и наоборот.
Кислота 1 + основание 2 основание 1 + кислота 2.
При этом, чем сильнее кислота, тем слабее сопряженное основание. Примером кислотно-основного катализа является гидролиз сложных эфиров.
+
СН3СООС2Н5 + Н2О →Н СН3СООН + С2Н5ОН Гомогенными также являются многие природные реакции,
катализируемые ферментами – биологическими катализаторами. Ферменты являются полимерами (белками) или комплексами полимеров с низкомолекулярными соединениями – активными центрами, которые и осуществляют превращения. Механизм их действия имеет свою специфику, например, включает образование комплекса активный центр фермента — реагент по типу «замок-ключ». Часто в состав активных центров входят металлы: Co, Ni, Fе (парфирины). Ферментативные катализаторы обычно выделяют в особый класс катализаторов. Многие из них значительно активнее неферментативных катализаторов. Например, фермент каталаза снижает энергию активации реакции разложения пероксида водорода в 10 раз, а скорость реакции увеличивает более чем на 10 порядков.
Гетерогенный катализ распространенное и широко используемое в промышленности явление. Каталитическое действие могут оказывать даже стенки реактора. Примеры каталитических реакций:
2H2+O2 |
Pt |
2H2O |
→ |
||
2KClO3 |
MnO |
2KCl+3O2 |
2 → |
||
CH3OH+O2 |
Cu |
CH2O+2H2O |
→ |
||
70% всех |
химических |
продуктов производится с использованием |
катализаторов. Крекинг нефти на алюмосиликатах, серная кислота – на оксиде ванадия, аммиак – на железе и т.д.
Для промышленных катализаторов важно, чтобы они имели большую площадь поверхности, на которой адсорбируются реагирующие молекулы. В промышленности используются катализаторы с удельной поверхностью 150 м2/г. Катализаторы на основе молекулярных сит (цеолиты, пористая керамика) имеют 1000 м2/г. Иногда вследствие спекания при высоких температурах поверхность уменьшается, поэтому применяют носители, которые препятствуют спеканию, и промоторы – вещества, небольшая добавка которых увеличивает каталитическую активность. Так в синтезе аммиака используют тонкодисперсное железо, осажденное на оксиде алюминия (носитель) с добавкой 1% оксида калия (промотор).
Участки поверхности, обладающие каталитической активностью, называются активными центрами. Они занимают небольшую часть поверхности. Что подтверждается явлением отравления катализаторов. Даже тысячные доли процента воды или оксида углерода (II) в азотоводородной смеси при синтезе аммиака, адсорбируясь на поверхности катализатора, резко
98
снижают его активность. Если поверхность железного катализатора покрыта на 10% водой, его активность снижается на 70%.
Вопрос о природе активных центров – предмет теории катализа. По Х.Тейлору – это группы атомов, выступающих над поверхностью. По мультиплетной теории Баландина молекулы реагирующих веществ взаимодействуют с несколькими атомами катализатора. Геометрия каталитического центра (расстояния между атомами, их взаимное расположение) предполагается близкой геометрии взаимодействующей молекулы. При этом происходит перераспределение электронной плотности химических связей реагирующей молекулы, что ведет к ее активации.
Кинетика гетерогенной химической реакции характеризуется наличием нескольких стадий:
1)перенос вещества из объема раствора к активной поверхности катализатора (внешняя диффузия);
2)диффузия по порам катализатора к внутренней поверхности гранул (внутренняя диффузия);
3)сама гетерогенная химическая реакция на поверхности, закономерности которой описываются кинетическими уравнениями.
В зависимости от того, какая стадия является самой медленной, говорят о кинетическом или диффузионном контроле.
Реакция, протекающая во внешнедиффузионной области, характеризуется: 1)зависимостью от интенсивности перемешивания жидкой фазы или скорости протекания газа через слой катализатора; 2)увеличением скорости реакции с уменьшением размера гранул катализатора; 3) энергией активации 4-8 кДж/моль, что соответствует энергии активации диффузии.
Если скорость реакции определяется скоростью внутренней диффузии, то 1)скорость реакции увеличивается с уменьшением размера гранул катализатора, при этом интенсивность перемешивания не влияет на скорость реакции; 2)порядок реакции изменяется с увеличением размера гранул при n>1; 3)скорость реакции зависит от пористости, радиуса гранул и извилистости пор.
Если реакция протекает в кинетической области, то ее скорость не зависит от перемешивания, размера гранул, пористости катализатора.
Переход из одной области в другую зависит от температуры, размера гранул: 1)при высоких температурах энергия активации низкая, наблюдается внешнедиффузионный контроль; 2)при низких температурах – кинетический контроль; 3) при средних – внутридиффузионный контроль.
Использование катализа в промышленности – это путь к высокоэффективным, ресурсосберегающим, малоотходным технологиям.
Экспериментальная часть
Определение кинетического порядка реакции.
Взаимодействие йодида калия с проксидом водорода в кислой среде происходит достаточно медленно, что дает возможность изучить кинетику этого процесса в условиях учебной лаборатории.
Сущность реакции окисления иодида калия пероксидом водорода сводится к взаимодействию ионов йода с молекулой пероксида водорода:
99
2KI + H2O2 +H2SO4 = I2 +2H2O + K2SO4 2I- +H2O2 = I2 + 2OH-
Эта стадия определяет скорость всей реакции, поскольку остальные стадии, являясь ионными реакциями, идут мгновенно. Реакция является тримолекулярной. В ходе опыта иодид-ион регенерируется добавлением тиосульфата натрия, таким образом, его концентрацию можно считать постоянной. Скорость реакции будет зависеть только от концентрации пероксида водорода, поэтому эту реакцию следует отнести к реакциям первого порядка.
Цель работы: ознакомиться с методом определения порядка реакции. Определить величину константы скорости реакции окисления иона йода пероксидом водорода и порядок этой реакции.
Оборудование и реактивы: коническая колба на 200 мл, штатив с бюреткой на 50 мл, мерные цилиндры на 100 мл и на 10 мл, пипетка на 10 мл, секундомер, раствор иодида калия (0,4%), раствор серной кислоты (2н), раствор пероксида водорода (0,05 н), раствор тиосульфата натрия (0,05 н), раствор крахмала (1%), раствор молибдата аммония (0,1 н).
Методика выполнения опыта. В коническую колбу на 200 мл отмерить цилиндром 100 мл 4%-ного (0,25 М) раствора иодида калия и 5 мл 2 н раствора серной кислоты. Над колбой укрепить бюретку с 0,05 н раствором тиосульфата натрия. Добавить в колбу из бюретки 1 мл раствора тиосульфата натрия, 15 капель крахмала, затем пипеткой внести 10 мл раствора пероксида водорода. Содержимое колбы тщательно перемешать.
Как только жидкость окрасится в голубой цвет, пустить в ход секундомер – это момент начала реакции. Секундомер не выключать до конца опыта.
Быстро прибавить из бюретки 1 мл раствора тиосульфата натрия, перемешать, окраска исчезнет. Время второго появления голубого окрашивания записать в таблицу (t1). В колбу прилить еще 1 мл тиосульфата натрия, записать время очередного появления окрашивания как t2. В общей сложности эту операцию повторять 5 раз, соответственно записывая время t1, t2, t3, t4, t5.
Затем в колбу прилить 5 капель 0,1 н раствора молибдата аммония, который является катализатором изучаемой реакции. Реакция мгновенно протекает до конца. Выделившийся йод оттитровать, добавляя по каплям из бюретки раствор тиосульфата натрия до исчезновения голубой окраски. Записать общий объем раствора тиосульфата, израсходованного в опыте (с). Этому количеству тиосульфата эквивалентны 10 мл 0,05 н раствора пероксида водорода.
Расчеты. В начальный момент опыта (первое появление голубого окрашивания) концентрация исходного вещества (а) соответствует (с-1) мл раствора тиосульфата натрия. К моменту второго появления окрашивания (t1) прореагирует еще порция раствора пероксида водорода эквивалентная 1 мл тиосульфата и , следовательно, х=1. К моменту t2: х=2 и т.д.
100