

1.
Энзимодиагностика-(энзим[ы] + греч. diagnostikos способный распознавать)
методы диагностики болезней, патологических состояний и процессов, основанные на определении активности энзимов (ферментов) в биологических жидкостях. В особую группу выделяются иммуноферментные диагностические методы, состоящие в применении антител, химически связанных с каким-либо ферментом, для определения в жидкостях веществ, образующих с данными антителами комплексы антиген — антитело.
Использование энзимных тестов является важным критерием в распознавании врожденных энзимопатий, характеризующихся специфическими нарушениями обмена веществ и жизнедеятельности в связи с отсутствием или недостатком того или иного фермента.
Ферменты представляют собой специфические высокомолекулярные белковые молекулы, являющиеся биологическими катализаторами, т.е. ускоряющими химические реакции, протекающие в живых организмах. Проникновение ферментов из клеток во внеклеточную жидкость, а затем в кровь, в мочу или другие биологические жидкости служит чрезвычайно чувствительным показателем повреждения плазматических мембран или повышения их проницаемости (например, вследствие гипоксии, гипогликемии, воздействия некоторых фармакологических веществ, инфекционных агентов, токсинов). Это обстоятельство лежит в основе диагностики повреждения клеток органов и тканей по феномену сопровождающей его гиперферментемии, причем выявляемое повышение активности фермента или его изоформы может иметь разную степень специфичности для поврежденного органа. Распределение отдельных изоферментов в тканях более специфично для определенной ткани, чем суммарная ферментативная активность, поэтому исследование некоторых изоферментов приобрело важное значение для ранней диагностики поражения отдельных органов и тканей. Так, например широко используется определение активности в крови изоферментов креатинфосфокиназы для диагностики острого инфаркта миокарда (Инфаркт миокарда), лактатдегидрогеназы — для диагностики поражений печени и сердца, кислой фосфатазы — и распознавании рака предстательной железы Диагностическая ценность энзимных тестов достаточно высока; она зависит как от специфичности данного вида гиперферментемии для определенных болезней, так и от степени чувствительности теста, т.е. кратности возрастания активности фермента при данном заболевании относительно нормальных значений. Однако большое значение имеет время постановки теста, т.к. появление и продолжительность гиперферментемии после повреждения органа различны и определяются соотношением скорости поступления фермента в кровоток и скорости его инактивации. При отдельных заболеваниях надежность их диагностики может быть повышена исследованием не одного, а нескольких изоферментов. Так, например, достоверность диагноза острого инфаркта миокарда возрастает, если в определенные сроки отмечено повышение активности креатинфосфокиназы, лактатдегидрогеназы и аспарагиновой аминотрансферазы. Степень выявляемой гиперферментемии объективно отражает тяжесть и распространенность повреждения органа, что позволяет прогнозировать течение заболевания.
2.
Митохондриа́льные заболева́ния — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими к нарушениям энергетических функций в клетках эукариотов, в частности — человека.
Митохондриальные заболевания обусловлены генетическими, структурными, биохимическими дефектами митохондрий, приводящими к нарушениям тканевого дыхания. Они передаются только по женской линии, к детям обоих полов, так как сперматозоиды переносят половину геномной информации, а яйцеклетка — поставляет и вторую половину генома, и митохондрии. Патологические нарушения клеточного энергетического обмена могут проявляться в виде дефектов различных звеньев в цикле Кребса, в дыхательной цепи, процессах бета-окисления и т. д.
Не все ферменты и другие регуляторы, необходимые для эффективного функционирования митохондрий кодируются митохондриальной ДНК. Большая часть митохондриальных функций контролируется ядерной ДНК.
Можно выделить 2 группы митохондриальных заболеваний:
Я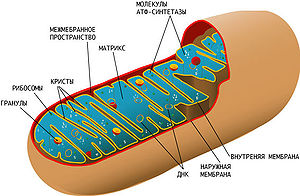 рко
выраженные наследственные синдромы,
обусловленные мутациями генов,
ответственных за митохондриальные
белки (синдром Барта, синдром Кернса-Сейра,
синдром Пирсона, синдром MELAS, синдром
MERRF и другие).
рко
выраженные наследственные синдромы,
обусловленные мутациями генов,
ответственных за митохондриальные
белки (синдром Барта, синдром Кернса-Сейра,
синдром Пирсона, синдром MELAS, синдром
MERRF и другие).
«Вторичные митохондриальные заболевания», включающие нарушение клеточного энергообмена как важное звено формирования патогенеза (болезни соединительной ткани, синдром хронической усталости, гликогеноз, кардиомиопатия, мигрень, печеночная недостаточность, панцитопения, а также гипопаратиреоз, диабет, рахит и другие).
Схема строения митохондрии. Сложная структура митохондрии и наличие собственной кольцевой хромосомы, кодирующей некоторые компоненты митохондрии, усложняет выяснение причин митохондриальных заболеваний
Дефекты и симптомы
Эффекты митоходриальных заболеваний очень разнообразны. Из-за различного распределения дефектных митохондрий в разных органах, мутация у одного человека может привести к заболеванию печени, а у другого — к заболеванию мозга. Величина проявления дефекта может быть большой или малой, и она может существенно изменяться, медленно наростая во времени. Некоторые небольшие дефекты приводят лишь к неспособности пациента выдерживать физическую нагрузку, соответствующую его возрасту и не сопровождаются серьёзными болезненными проявлениями. Другие дефекты могут быть более опасны, приводя к серьёзной патологии.
В общем случае митоходриальные заболевания проявляются сильнее при локализации дефектных митохондрий в мышцах, мозге, нервной ткани, поскольку эти органы требуют больше всего энергии для выполнения соответствующих функций.
Несмотря на то, что протекание митохондриальных заболеваний сильно отличаются у разных пациентов, на основании общих симптомов и конкретных мутаций, вызывающих болезнь, выделено несколько основных классов этих заболеваний.
Типы заболеваний
Помимо относительно распространённой митохондриальной миопатии, встречаются:
Сахарный диабет, сопровождающийся глухотой (DAD, MIDD): Это сочетание в раннем возрасте может быть вызвано мутацией митохондриального гена MT-TL1, но сахарный диабет и глухота могут быть вызваны как митохондриальными заболеваниями, так и иными причинами
наследственная оптическая нейропатия Лебера (en:Leber's hereditary optic neuropathy (LHON)), характеризующийся потерей зрения в раннем пубертатном периоде;
синдром Вольфа-Паркинсона-Уайта (У части больных может не выявляться клинических проявлений. Основное проявление синдрома Вольфа-Паркинсона-Уайта — аритмии. Более чем в 50 % случаев возникают пароксизмальные тахиаритмии: наджелудочковые реципрокные, фибрилляция предсердий, трепетание предсердий. Довольно часто синдром возникает при заболеваниях сердца — аномалии Эбштайна, гипертрофической кардиомиопатии, пролапсе митрального клапана.)
рассеянный склероз и подобные ему заболевания;
синдром Лея (Leigh) или подострая некротизирующая энцефаломиопатия : После начала нормального постнатального развития организма болезнь обычно развивается в конце первого года жизни, но иногда проявляется у взрослых. Болезнь сопровождается быстрой потерей функций организма и характеризуется судорогами, нарушенным состоянием сознания, деменцией, остановкой дыхания
«Нейропатия, атаксия, retinitis pigmentos и птоз» en:Neuropathy, ataxia, retinitis pigmentosa, and ptosis (NARP): прогрессирующие симптомы нейропатии, атаксии, тунельное зрение и потеря зрения, птоз, деменция;
Митохондриальная нейрогастроинтенстинальная энцефалопатия en:Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): гастроинтестинальная псевдообструкция и кахексией, нейропатия, энцефалопатия с изменениями белого вещества головного мозга
Диагностика
Для постановки диагноза митохондриального заболевания важен комплексный генеалогический, клинический, биохимический, морфологический и генетический анализ.
У некоторых больных с характерной картиной перечисленных выше синдромов диагноз может быть подтвержден молекулярно-генетическим тестированием ДНК, полученной из крови больного.В других случаях диагноз возможен на основе родословной, концентрации лактата в спиномозговой жидкости (СМЖ) и в крови, МРТ головного мозга, ЭКГ, биопсии мышц с гистологическим и гистохимическим исследованием, молекулярно-генетических исследований мутации мтДНК.
Эпидемиология
Изначально мутации мтДНК считались крайне редкими, однако исследование 3000 здоровых новорожденных на 10 наиболее известных патогенных мутаций, проведённое в 2008 году, выявило таковые у одного человека из 200.[3] "Горячей точкой" в мтДНК оказалась позиция 3243, здесь часто происходит замена A-G, изменяющая функционирование гена MT-TL1.
Лечение
В настоящее время лечение митохондриальных заболеваний находится в стадии разработки, но распространённым терапевтическим методом служит симптоматическая профилактика с помощью витаминов. Также в качестве одного из методов применяются пируваты
Поскольку в сперматозоиде, который вносит половину хромосом будущего организма, содержится мало митохондрий, митохондриальная наследственность определяется, в основном, митохондриями яйцеклетки. Сейчас проводятся экспериментальные работы по экстракорпоральному (in vitro) оплодотворению с использованием переноса ядра оплодотворённой яйцеклетки в безъядерную цитоплазму другой яйцеклетки с нормально функционирующими митохондриями (замена ядра).
3.
I. Нарушения метаболизма аминокислот
1.1.Нарушения метаболизма гли.
Нетоксическая гиперглицинемия – причиной является дефицит ТГФК- редуктазы в реакции:
ГЛИ + НАД + ТГФК СО2 + метилен-ТГФК + НАДН2.
Дефицит фермента или полное отсутствие в ткани мозга- является причиной гиперглицинемии. Существует четкая зависимость между остаточной активностью фермента и выживаемостью больных.
У новорожденных отсутствует реакция на окружение, икота, гипотрофия и клонические судороги. У детей старшего возраста - задержка умственного развития и судороги. Содержание ГЛИ в моче выше нормы в 10-20 раз, а в крови в 3-5 раз при отсутствии накопления других аминокислот.
1.2.Нарушения метаболизма цис
Наследственные нарушения обмена серосодержащих АмК.
Ц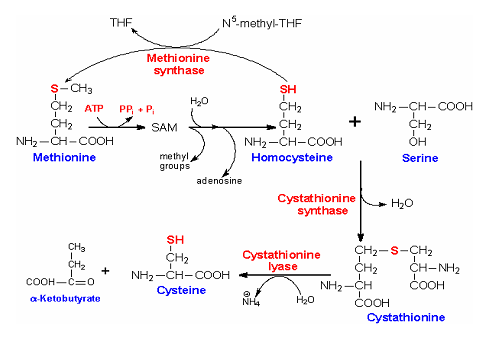 истинурия,
цистиноз распространенное врожденное
заболевание. Встречается с частотой (
1: 600), уступая только ФПО. Нарушение
метаболизма Метеонина связано со
снижением или отсутствием активности
фермента цистатионин- синтетазы
истинурия,
цистиноз распространенное врожденное
заболевание. Встречается с частотой (
1: 600), уступая только ФПО. Нарушение
метаболизма Метеонина связано со
снижением или отсутствием активности
фермента цистатионин- синтетазы
Цистинурия- это аномалия обмена, связанная с образованием камней в почках, мочевом пузыре, мочеточниках. Как следствие отложение кристаллов цистина, на фоне глюкозурии, фосфатурии, общей аминоацидурии. Гомоцистинурия генетически гетерогенна. Полиморфизм проявляется в следующих формах:
1. Подвывиха хрусталика. Признаком является- эктопия хрусталиков- дрожание радужки при быстрых движениях головы ребенка. Нарушение связано с утолщением и фрагментацией зонулярных волокон, крепящих хрусталик к реснитчатому телу. Кроме того возможен остеопороз, вальгусное искривление голени, полая стопа. В половине случаев- УО, у 10-15% - судороги или тромбоэмболия.
2.Гетерогенная форма связанна с нарушением использования витамина В6.
Гомоцистеин - продукт, лежащий на перекрестке всех превращений МЕТ. Соединяясь с СЕР- образует Цистатионин, или реметилируясь, он снова превращается в МЕТ. Дефицит цистатионинсинтетазы блокирует синтез цистатионина, и сопровождается усиленным превращением МЕТ в гомоцистеин. Последний тормозит образование поперечных сшивок в коллагене, блокирует активные группы ЛИЗ и окси-ЛИЗ, которые образуют поперечные сшивки. При отсутствии последних, с возрастом происходит накопление гомоцистеина, что можно расценивать как риск-фактор в развитии многих заболеваний. При гомоцистинурии в патологический процесс вовлекается множество органов и систем. Смерть наступает рано от тромбоза крупных артерий. При гомоцистинурии повышено выделение с мочой оксипролина и кислых ГАГ. В крови в 4-5 раз повышен МЕТ. В моче геперметеонинурия и гомоцистинурия.
3.Нарушение метаболизма фолиевой и ТГФК сопровождается мышечной адинамией. При отсутствии последних, с возрастом происходит накопление гомоцистеина, что можно расценивать как риск-фактор в развитии многих заболеваний.
Формы цистинурии
Признаки |
Причины, дефект фермента
Цистатионин Гомоцис-СН3-трансфер СН3-ТГФК-редук-за
си-
|
Задержка развития |
Часто Часто |
Подвывих хрусталика |
Почти всегда Нет |
МЕТ в крови |
Выше нормы Норма или ниже |
Гомоцис в крови и моче |
То же Выше нормы |
Цистатионин в крои и моче |
Ниже нормы Норма или выше |
Реакция на ограничение Мет в пище |
Положительная Отрицательная |