

1.4. Нарушения метаболизма трп
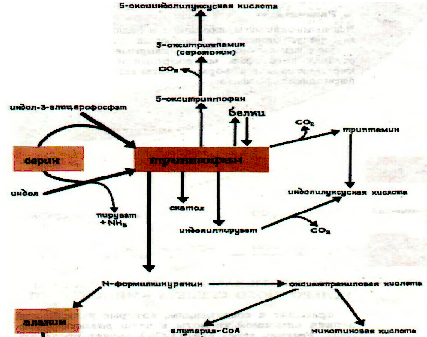
Первичные нарушения обмена связаны с генетическими факторами, и проявляются в виде:
Нарушении всасывания ТРП- болезнь Гартнупа
Ферментативных блоков в метаболизме ТРП – синдром « Голубых пеленок»
Синдрома-Тада
Синдрома Прайса
Наследственной ксантуренурии.
Вторичные нарушения метаболизма ТРП зависят от гормонального статуса и обеспеченности витаминами, особенно В6.
1.5. Нарушения метаболизма гис
Гистидинемия- дефект гистидазы ( гистидин-NH2-лиазы), катализирующей переход ГИС в 5- формимино ТГФК. Реакция протекает параллельно с декарбоксилированием ГИС. Клинически проявляется дефектами речи у ребенка, нарушением слуховой памяти, снижением интеллекта, частыми инфекционными заболеваниями, а также малым ростом ребенка.
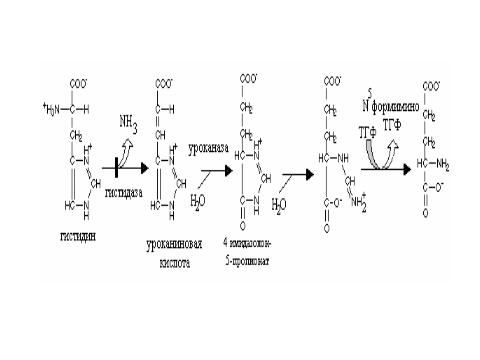
Гистидин-аммиак – лиаза печени, это фермент, постоянно востребующий группы SH.
Фермент зависим от влияния гипофизарных гормонов. При гипофизэктомии ( в эксперименте) повышает его активность. У лиц с врожденной гистидинемией фермент отсутствует вообще. В крови и моче ГИС повышен.
Уроконатгидратаза катализирует трансформацию в имидазолон-3-пропионат. Реакция требует включения ТГФК при участии специфической трансферазы. Атом С2- имидазольного кольца гистидина возвращается в пул одноуглеродных фрагментов. Животные и люди с недостатком ФК или витаминов В12 выделяют большое количество формиминоглутаминовой кислоты.
Главный путь метаболизма формиминогруппы -это ее превращение в СН3.
Неспецифическая декарбоксилаза ароматических аминокислот имеет низкую активность по отношению к Гистидину. Специф. фермент, имеющийся в тучных клетках, основном месте образования гистамина, является ся главным катализатором декарбоксилирования ГИС. Во многих др. тканях- печени, легких, мышцах и слизистой желудка, содержание этого амина высокое , вследствие его синтеза in situ.
Гистамин- мощный сосудорасширяющий агент и при достаточно высоких концентрациях может вызвать сосудистый коллапс. Образуется при травматическом шоке и в зоне воспалительного процесса. Стимулирует в желудке секреции как пепсина, так и кислоты. Применяется для исследования функции желудка. Диаминооксидаза превращает ГИС в соответствующий альдегид и NH3. Часть неразрушенного ГИС экскретируется с мочой в виде N- ацетильного и N - метильного производного ( главный метаболит Гис у человека).
Стойкая гипер ЛИЗинемия- связана со значительным снижением активности лизин -альфа- кето-ГЛУ- редуктазы, которая катализирует 2 реакции:
Соединение ЛИЗ с альфа- кето-ГЛу- образование САхаропина;
Редукция Сахаропина- появление аминоадипиновой кислоты.
Типичными являются проявления УО, аутизм, низкорослость, глухота, судороги, разболтанность суставов.
4.
Энзимопатии синтеза пуринового кольца
Основной фонд нуклеотидов создается путем синтеза de novo из простых предшественников
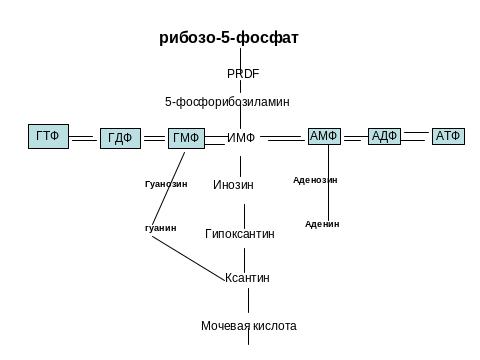
Гиперурикемия- повышение концентрации мочевой кислоты в плазме крови. Причины:
-Увеличение скорости синтеза пуринов;
-Ускоренный катаболизм НК( при заболеваниях, сопровождающихся клеточной
пролиферацией; псориаз, рак, лимфома, лейкемия);
-Снижение уровня экскреции.
Наиболее распространенное заболевание- подагра.
Одно из характерных проявлений П- подагрический артрит
Дефектный ф-т-ФРДФ-синтетаза, проявляется гиперурикемией, подагрой
GGPRT- клинически те же проявления при частичной потере активности фермента. При полной потере активности GGGPRT- болезнь Леша- Нихана
АДА- потеря активности АДА –проявляется как комбинированный (Т и В клетки) иммунодефицит, гипоурикемией, дезоксиаденозинурией.
ПНФ- при потере активности- иммунодефицит (Т клетки).
Аденозинфосфорибозилтрансфераза- при полной потере активности почечно-каменная болезнь
Ксантиноксидаза при полной потере активности- гипоурикемия, ксантиновые камни в почках.
Ферменты обмена пуриновых нуклеотидов в функционировании Т и В лимфоцитов и в патогенез иммунодефицитов
Аденозиндезаминаза (АДА) или аденозинаминогидролаза (КФ 3.5.4.4) ключевой фермент катаболизма пуриновых нуклотидов, обмен которых играет важную роль в функционировании разнообразнвх клеток организма человека. Особенно важен метаболизм нуклеотидов в реализации функций клеток иммунной системы: Т и В лимфоцитов ( созревание, апоптоз, иммунный ответ и при нарушениях ( лейкозы, первичные и вторичные иммунодефициты).
Дезоксиаденозин + Н2О ----- Дезоксиинозин +NH3
Аденозин +Н2О --------- Инозин +NH3
Фермент имеет относительную специфичность, катализирует дезаминирование различных производных аденина, часть которых яв-ся лекарственными препаратами ( противоопухолевыми, противовирусными). Изучены 2 изофермента АДА с различным распределением в лимфоидных и нелимфоидных
клетках и с разной чувствительностью к ингибиторам АДА, применяемым для лечения лейкозов.
При генетическом дефекте АДА у детей в период до 2 лет проявляется тяжелый комбинированный иммунодефицит, связанный с нарушением созревания и пролиферации Т и В лимфоцитов, лимфопенией, слабым иммунным ответом на антиген, пониженным уровнем иммуноглобулинов в крови и повышенным содержанием аденозина и дезоксиаденозина. Дефект АДА проявляется во многих тканях, но патолог. последствия развиваются в лимфоцитах. Недоразвиты тимус и лимфатические узлы. Дети предрасположены к бактериальным, вирусным и грибковым заболеваниям. Обычно умирают к 2-летнему возрасту в результате повреждения бронхолегочной системы, дисплазии костного мозга и др. причин.
Наследственный дефицит АДА связан с рецессивными мутациями ( замены и делеции нуклеотидов) в гене АДА, в 20 хромосоме человека.
Механизм токсических и иммуносупрессивных эффектов при наследственном дефекте АДА связан с торможением р-ции дезаминирования и приводит к увеличению концентрации Аденозина и дезоксиаденозина в клетках и крови. Поэтому повышенно содержание адениловых и дезоксиадениловых нуклеотидов и даже цАМФ. Дезоксиаденозин и особенно dATP токсичны для лимфоцитов, угнетают рибонуклеотидредуктазу и снижают синтез dNTF и ДНК, В др. тканях в отличие от лимфоцитов активна фосфатаза dATF, которая предотвращает накопление dATF и последствия угнетения или дефицита АДА. Торможение образования дезоксиинозина и инозина и далее гипоксантина, снижает продукции АФК, которые нужны для нормального функционирования лейкоцитов и лимфоцитов. АФК накапливаются в результате побочных реакций, катализируемых ксантиоксидазой.
Для лечения комбинированного иммунодефицита может быть использована пересадка костного мозга при наличии здоровых доноров.
Другой важный фермент катаболизма пуриновых нуклеотидов- пуриннуклеотидфосфорилаза (ПНФ) или ортофосфат рибозилтрансфераза (КФ 2.4.2.1). Катализирует реакции с эндогенными субстратами:
1. дезоксигуанозин + Н3РО4 ----- гуанин + 2/- дезоксирибоозо-1-фосфат;
2. гуанозин + Н3РО4 ------ гуанин + рибозо-1 –фосфат;
3. инозин + Н3РО4 ------ гипоксантин + рибозо-1-фосфат.
Фермент активен во многих клетках, но наиболее существенны его функции для Т- лимфоцитов. Интенсивность ПНФ особенно высока в незрелых тимоцитах и снижается в периферических Т- лимфоцитах.
При врожденном дефекте ПНФ у детей во втором полугодии жизни или позже, проявляется предрасположенность к различным инфекционным заболеваниям, связанным с ослаблением иммунитета. Нарушается созревание Т-лимфоцитов, снижается их содержание в крови. При этом содержание В- лимфоцитов и иммуноглобулинов в крови существенно не меняется. В клетках и в крови повышены концентрации субстратов ПНФ и конц. токсического dGTF в Т- лимфоцитах и даже эритроцитах ( что может быть причиной анемии). Дефицит ПНФ проявляется в виде более легких заболеваний, чем дефицит АДА.
Т.о. наследственный дефект ПНФ снижает количество или изменяет функции Т- лимфоцитов, что приводит к нарушению Т- опосредованного( клеточного) компонента иммунитета.
В настоящее время в практике трансплантологии внедрены иммунодепрессанты- ингибиторы инозинмонофосфат-ДГ лимфоцитов, катализирующей одну из р-й синтеза GMР. ( Препараты мизорбин, брединин т.д.).
5.
Принципиально различны ферменты карбомоилфосфат синтаза- I , и карбм.ф-т синтаза- II, локализованные в различных отделах клетки, контролирующие разные процессы.
Первый отвечает за синтез мочевины, локализован в Мит., второй контролирует б/з пиримидинов в цитоплазме.
Дефект карбм. фосф. син-зы I, яв-ся аутосомно- рецессивным, проявляется в течении 24-48 час после рождения ребенка. развивается гипераммониемия и наступает смерть. В крови повышены ГЛУ и АЛА. В моче Оротат. Лечение основано на гемодиализе, снижении количества белков в пище, введении бензоата, фенилацетата.
Дефект орнитинкарбомоилфосфаттрансферазы сцеплен с Х хромосомой. Клинически проявляется гипераммониемией, летаргией, гипотонией, снижением толерантности к белкам В крови увеличены ГЛУ и АЛА. В моче повышен Оротат. При низкой активности I, с мочой выделяются оротат, уридин и урацил, которые, синтезируются в цитозоле при участии карб.синтаза-II в избытке субстратов, не используемых для синтеза М.
Дефект аргининосукцинат синтетазы - приводит к цитруллинемии. Избыток Цитруллина в крови, моче, цереброспинальной жидкости может превышать норму более, чем в 100 раз. Тип наследования- аутосомно- рецессивный. Гипераммониемия очень тяжелая у новорожденных. У взрослых людей после белковой нагрузки. Цитруллин повышен в крови и моче. Лечение основано на ограничении белков и введении АРГ и ГЛУ. Эта реакция важна, т.к. в ней связывается второй атом азота, вводимый в молекулу М за счет АСП.
Дефект аргининосукцинатслиазы возникает по аутосомно-рецессивному признаку, и проявляется в клинике гипераммониемией, атаксией, выпадением волос. В крови и моче появляется аргининосукцинат, ГЛН, АЛА. ЛИЗ. Потребление белков следует ограничить.
Ферментативная несостоятельгость аргиназы( аутосомно- рецессивный признак) проявляется гипераргининемией. В клинике отмечается повышенный АРГ в крови и моче. Белки должны быть ограниченны.
При различных дефектах ОРН –цикла, лечение направлен на снижение концентрации аммиака за счет малобелковой диеты, стимуляции его( аммиака) выведения в обход нарушенных реакций.
Это достигается двумя путями:
Связыванием и выведением аммиака в обход ОРН цикла в составе фенилацетилглутамина и гиппуровой кислоты;
Повышением концентрации промежуточных метаболитов цикла, образующихся вне блокируемых реакций.
Бензойная кислота в печени быстро метаболизируется в гиппуровую к-ту в реакции с глицинацилазой. Гиппурат бысто выводится с мочой. ГЛН восстанавливается из 2х источников:
из СЕР и за счет его синтеза из аммиака и СО2 в р-ции с ГЛН_синтазой. В обоих случаях на синтез ГЛИ используется одна молекула аммиака. Эта тактика стимуляции интактных р-ций выведением метаболитов ОРН. цикла основана на том, что промежуточные продукты ЦСМ менее токсичны, чем свободный аммиак. В различных комбинациях эти методы влияют на продолжительность жизни больных с дефектами ферментов в ЦСМ, хотя не могут полностью предотвратить изменения в ЦНС.
Для предотвращения нейротоксического действия аммиака требуется ранняя диагностика.
Токсичность аммиак. Повышенный уровеньаммиака в плазме крови снижает фонд метаболитов и АТФ в клетках ЦНС.
Альфа-кето-ГЛУ + NH3 + NADFH+H ----- NADF + ГЛУ
ГЛУ + NH3 +АТФ ------ ГЛН + АДФ + Н3РО4
Накопление ГЛН в клетках нейроглии приводит к повышению в них осмотического давлении и набуханию астроцитов.
При рН 7.4 аммиак существует в крови почти целиком в виде катиона
аммония. Его ионы проходят через плазматическую и митохондриальную мембраны с большим трудом. Повышение его концентр. создает препятствия для движения других ионов и приводит к сдвигу рН. При концентрации в 1% он легко проходит через мембраны клеток и в митохондриях сдвигает равновесие ГЛ- ДГ –реакции в сторону образования ГЛН. Это нарушает процесс синтеза тормозного медиатора ЦНС- гамма- аминомасляной к-ты.
Кроме того, нарушение метаболизма Амк. в печени приводит к повышению конц. в крови циклических аминокислот и ЖК с короткой цепью, что оказывает токсическое влияние на клетки мозга.
Гипераммониеми я может возникать при различных заболеваниях печени: цирроз, жировая дистрофия, гепатит.
Патология обмена белков – материал достаточно известный.
8.
Нарушения обмена фруктозы и галактозы
Эссенциальная фруктозурия обусловлена недостаточностью фосфофруктокиназы, фосфорилирующей ее по первому атому. Это исключает поступление фруктозы в клетки, так как моносахариды внутри клеток находятся только в осфорилированном состоянии. Уровень фруктозы в крови повышается, и она появляется в моче. Клинически болезнь не проявляется, выявляется случайно при исследовании мочи на сахар. Для исключения сахарного диабета проводят реакцию Селиванова на фруктозу.
Положительная реакция указывает на фруктозурию.
Наследственная непереносимость фруктозы клинически проявляется при включении в рацион ребенка фруктов или соков. Признаки: рвота, боли в животе, диарея, гипогликемия. В основе – недостаточность фруктозо-1-фосфатальдолазы. Накопление фруктозо-1-фосфата приводит к торможению гликогенолиза, так как он ингибирует гликогенфосфорилазу, а также метаболизма глюкозы из-за ингибирования фосфогексоизомеразы. Это уменьшает скорость утилизации глюкозы и производства АТФ, вызывает гипофосфатемию и служит сигналом для расщепления адениловых нуклеотидов – источников внутриклеточного фосфора. В диагностике наследственной непереносимости фруктозы особое значение имеет выяснение связи между приемами фруктозосодержащей пищи и приступами заболевания. Из лабораторных данных наиболее важны: выявление в моче редуцирующих веществ и их дифферециация (фруктоза или галактоза); результаты пробы на олерантность к фруктозе (прием 0,25г на 1 кг веса тела). Наследственная непереносимость фруктозы – довольно часто встречающееся заболевание, потому что может быть также обусловлена дефицитом фруктозо-1,6-дифосфатазы. Механизм развития биохимических сдвигов примерно тот же, что и при недостатке фруктозо-1-фосфатальдолазы.
Особое место среди патологий углеводного обмена занимает галактоземия. У больных в крови обнаруживается большое количество галактозы и галактозо-1-фосфата. Проявляется галактоземия в раннем возрасте и часто приводит к гибели детей, так как поражаются почки, печень, ЦНС, развивается катаракта. Сопровождается галактозурией, выделением аминокислот с мочой. В основе заболевания – нарушение ферментативных превращений галактозы в глюкозу. Метаболические ревращения галактозы в клетках осуществляются по следующему пути:
Галактоза + АТФ------ Галактозо-1-фосфат + АДФ
Галактокиназа
Галактозо-1-фосфат+УДФ-глюкоза→УДФ-галактоза+Глюкозо-1-фосфат
Галактозо-1-фосфат-уридилтрансфераза
УДФ-галактоза---------- УДФ-глюкоза
Эпимераза
Дефицит галактокиназы приводит к накоплению избытка галактозы, не вовлекаемой в метаболические процессы, и dыведению ее с мочой. Заболевание проявляется ухудшением зрения и развитием катаракты. Это связано с увеличением вероятности восстановления галактозы при участии НАДФН в спирт (галактитол). Накапливаясь в стекловидном теле, галактитол связывает большие количества воды, что сопровождается разрывом зонулярных волокон. Лабораторными показателями недостаточности галактокиназы служат снижение активности фермента в эритроцитах и наличие галактозы в моче.
Дефицит галактозо-1-фосфат-уридилтрансферазы сопровождается накоплением и галактозы, и галактозо-1-фосфата. Недостаточность проявляется сразу после начала кормления в виде рвоты, диареи, желтушности сохраняющейся более длительно, чем обычно. Биохимические изменения включают галактоземию, галактозо-1-фосфатемию, аминоацидурию, про-
теинурию. Лабораторная диагностика: снижение активности фермента в эритроцитах (от 10 до 50% от нормы при разных формах заболевания). Рекомендуется исключение продуктов, содержащих галактозу из пищи, переход на кормление смесями, не содержащими лактозу. Дефицит уридинфосфатгалактозо-4-эпимеразы – редкое явление, описан лишь у больных одной семьи: наблюдали гипергалактоземию и галактозурию при отсутствии каких-либо клинических проявлений.
9.
Гемолитические анемии. ДЕФИЦИТ ГЛЮКОЗО-6-ФОСФАТДЕГИДРОГЕНАЗЫ И ДРУГИХ ФЕРМЕНТОВ ЭРИТРОЦИТОВ.
Наиболее распространённый вариант энзимопатий, приводящий к развитию гемолитической анемии — недостаточность глюкозо-6-фосфат дегидрогеназы. Заболевание широко распространено среди афроамериканцев (6–30%), меньше — среди татар (3,3%), народностей Дагестана (5–11,3%); в русской популяции выявляют редко (0,4%). Частный случай недостаточности глюкозо-6-фосфат дегидрогеназы — фавизм. Гемолиз развивается при употреблении в пищу конских бобов, фасоли, гороха, вдыхании нафталиновой пыли.
Этиология и патогенез. Наследование недостаточности глюкозо-6-фосфат дегидрогеназы (К), в силу чего чаще болеют мужчины. В мире насчитывают около 400 млн носителей этого патологического гена. Заболевание развивается, как правило, после приёма определённых лекарственных средств [производные нитрофурана, хинин, изониа^ид, фтивазид, аминосалициловая кислота (натрия пара-аминосалицилат), налидиксовая кислота, сульфаниламиды и др.] или на фоне инфекции.
Клиническая картина. Заболевание проявляется бурным развитием гемолиза при употреблении перечисленных выше веществ или инфекциях (особенно при пневмониях, брюшном тифе, гепатите). Недостаточность глкжо-зо-6-фосфат дегидрогеназы может быть причиной желтухи новорождённых. В анализе крови выявляют ретикулоцитоз, повышение уровня прямого и непрямого билирубина, ЛДГ, щелочной фосфатазы. Морфология эритроцитов и эритроцитарные индексы не изменены. Диагноз устанавливают на основании результатов определения активности фермента.
Лечение. Вне криза лечение не проводят. При лихорадке применяют физические методы охлаждения. При хроническом гемолизе назначают фолиевую кислоту 1 мТ/сут по 3 нед каждые 3 мес. При кризе отменяют все лекарственные средства, проводят инфузионную терапию на фоне дегидратации.
Глюкозо-6-фосфатдегидрогеназа (Г-6-ФДГ) катализирует начальный этап в пентозофосфатном пути гликолиза. Главной функцией пентозофосфатного пути является превращение никотинамид аденин динуклеотид фосфата (НАДФ) в НАДФН.
Для превращения оксидированного глютатиона в редуцированную форму необходим НАДФН. Редуцированный глютатион выполняет функцию детоксикации перекиси водорода в органические перекиси.
ПАТОГЕНЕЗ ГЕМОЛИЗА ЭРИТРОЦИТОВ ПРИ ДЕФИЦИТЕ Г-6-ФДГ
Принципиальный базис разрушения эритроцитов, дефицитных по Г-6-ФДГ, до настоящего времени полностью неясен и, возможно, имеет различия при разных гемолитических синдромах.
Дефицитные по Г-6-ФДГ клетки ограничены в своей способности генерировать НАДФН и затем образовывать редуцированную форму глютатиона, которая необходима для уменьшения содержания перекиси водорода и свободных радикалов, возникающих при функционировании клетки.
Кислородный «взрыв», возникающий вследствие избытка перекиси водорода и других форм активированного кислорода, ведет к денатурации белка, который прикрепляется к мембране эритроцита (тельца Гейнца) и способствует изменению как формы, так и структуры мембраны эритроцита.
При прохождении эритроцитов через печень и селезенку тельца Гейнца вместе с частью клеточной мембраны «отщипываются» макрофагами.
КЛИНИЧЕСКАЯ КАРТИНА
Клиническая манифестация наследственного дефицита фермента эритроцитов складывается из:
эпизодов гемолиза после воздействия оксидантов или инфекции
хронической гемолитической анемии (наследственная несфероцитарная анемия)
острого гемолиза после употребления бобов («фавизм»)
метгемоглобинопатии
желтухи новорожденных
ОСТРАЯ ГЕМОЛИТИЧЕСКАЯ АНЕМИЯ
Лекарственный гемолиз обычно начинается через 2-4 дня после приема лекарственного препарата. Симптомы и объективные признаки острого гемолиза:
желтуха
бледность
темная моча с появлением (или без них) болей в спине и животе
концентрация Hb может достигать 30-40 г/л
в мазке крови выявляют фрагментацию эритроцитов и эритроциты с отщепленной частью мембраны (дегмациты)
Острый гемолиз заканчивается спонтанно через 1 неделю, концентрация Hb медленно возвращается к нормальному уровню.
Лекарственные препараты, вызывающие гемолиз при дефиците Г-6-ФДГ:
Противомалярийные препараты:
Примаквик
Пентаквин
Сульфаниламиды:
Сульфазин
Сульфален
Сульфапиридин
Сульфаметоксазол
Сульфоны:
Тиазолисульфон
Диафенилсульфон (дапсон)
Нитрофураны:
Фуразолидон
Анальгетики:
Ацетиламид
Неграм
Химические вещества:
Метиленовый синий
Нафталин
Фенилгидразин
Толуидин синий
Тринитротолуол
ГЕМОЛИЗ, ИНДУЦИРОВАННЫЙ ИНФЕКЦИЕЙ
Инфекции являются более частой причиной развития гемолиза при дефиците Г-6-ФДГ.
Большое количество инфекционных агентов могут провоцировать возникновение гемолиза: сальмонеллы, b-гемолитический стрептококк, риккетсии. Особенно выраженный гемолиз наблюдается при вирусных гепатитах.
Гемолиз обычно умеренный, почечная недостаточность развивается вторично по отношению к массивному внутрисосудистому гемолизу.
Желтуха может быть выраженной при сочетании с вирусным гепатитом.
Ретикулоцитоз обычно отсутствует, выздоровление от анемии зависит от успешной терапии основного заболевания.
ГЕМОЛИЗ ПРИ ДИАБЕТИЧЕСКОМ АЦИДОЗЕ
Диабетический ацидоз может вызвать умеренный гемолиз эритроцитов, дефицитных по Г-6-ФДГ.
Коррекция ацидоза и введение глюкозы прекращают гемолитический процесс.
НЕОНАТАЛЬНАЯ ГИПЕРБИЛИРУБИНЕМИЯ
Появляется у некоторых новорожденных с дефицитом Г-6-ФДГ без признаков иммунизации матери фетальным Hb.
Желтуха новорожденных приводит к развитию поражения центральной нервной системы и умственной отсталость.
НАСЛЕДСТВЕННАЯ НЕСФЕРОЦИТАРНАЯ ГЕМОЛИТИЧЕСКАЯ АНЕМИЯ
При некоторых вариантах дефицита Г-6-ФДГ наблюдается гемолиз эритроцитов при отсутствии воздействия провоцирующих лекарственных препаратов или инфекции.
Анемия и желтуха чаще всего диагностируются в неонатальном периоде. Гипербилирубинемия может потребовать обменных трансфузий.
Анемия варьирует от тяжелой (уровень Hb 50 г/л) до полностью компенсированного статуса с нормальной концентрацией Hb.
Около 50 % всех эритроцитов имеют значительно укороченную продолжительность жизни (от 2 до 17 дней). Наблюдается нарушение функции нейтрофилов в отношении киллинга микроорганизмов при сохранном хемотаксисе и поглотительной способности этих клеток.
Характерны: хроническая желтуха, спленомегалия, желчнокаменная болезнь, язвы кожи лодыжек.
Негематологические проявления дефицита Г-6-ФДГ: катаракта и отложение гликогена при дефиците фосфофруктокиназы.
Часто наблюдаются рецидивирующие инфекции, вызванные каталазо-позитивными микроорганизмами.
Спленэктомия неэффективна.
ФАВИЗМ
Один из наиболее тяжелых проявлений дефицита Г-6-ФДГ. Характерен для детей в возрасте от 1 до 5 лет.
Острый гемолиз появляется через 5-24 часа после приема бобов.
Пациенты предъявляют жалобы на головную боль, тошноту, боли в спине, кожные высыпания, лихорадку, которые обычно появляются вслед за гемоглобинурией, анемией и желтухой.
Происходит значительное снижение концентрации Hb (до 60 г/л у 80 % и ниже 40 г/л — у 30 % пациентов).
Моча становится красной или темной, может развиться шок при массивном внутрисосудистом гемолизе (опасность развития ОПН). Вследствие невозможности проведения трансфузионной терапии смертность при фавизме составляет около 8 %.
После 3-4-х дней от начала гемолиза происходит медленное выздоровление.
ДЕФИЦИТ ДРУГИХ ФЕРМЕНТОВ
Дефицит g-глютамил-цистеин синтетазы.
Наследуется по аутосомно-рецессивному типу.
Характеризуется умеренной гемолитической анемией. Осмотическая резистентность эритроцитов нормальная, слегка повышен аутогемолиз.
ДЕФИЦИТ ГЛЮТАТИОН-СИНТЕТАЗЫ
Характеризуется умеренной гемолитической анемией, тяжелым метаболическим ацидозом, сочетающимся с 5-оксипролинурией, желтухой и нарушением умственных способностей.
Прием оксидантных препаратов способствует усилению гемолиза.
Кровь: анемия, ретикулоцитоз, анизо-, пойкилоцитоз, наличие телец Гейнца, положительный цианид-аскорбиновый тест.
При более генерализованном синдроме развиваются: прогрессирующая церебральная и мозжечковая дегенерация, атрофия гранулярных клеток мозжечка и фокальные поражения коры и таламуса.
Спленэктомия дает частичное улучшение. Метаболический ацидоз успешно лечится постоянным назначением щелочных растворов.
ДЕФИЦИТ ГЛЮТАТИОН ПЕРОКСИДАЗЫ
Фермент содержит селен как интегрирующую часть своей структуры, поэтому недостаток микроэлемента приводит к снижению активности глютатион пероксидазы.
Дефицит активности глютатион пероксидазы может развиться при дефиците железа, циррозе печени, тромбоцитопатии Гланцмана, сульфметгемоглобинемии, a-талассемии.
Клинически дефицит фермента протекает с незначительными признаками гемолитической анемии.
ДЕФИЦИТ ГЛЮТАТИОН S-ТРАНСФЕРАЗЫ
Энзим играет роль в активном транспорте глютатион S-трансферазно-ксенобиотических конъюгатов из эритроцитов.
Изолированный дефицит описан у мужчин с умеренной гемолитической анемией и спленомегалией.
10.
Теория адекватного питания, ее основные постулаты.
Кризис теории сбалансированного питания привел к пересмотру ряда основных ее положений и формированию на ее основе новой теории адекватного питания. Вот ее основные постулаты:
Питание поддерживает молекулярный состав и возмещает энергетические и пластические расходы организма на основной обмен, рост и выполненную работу. (Этот постулат единственный общий с классической теорией).
Необходимыми компонентами пищи являются не только нутриенты, но и балластные вещества (пищевые волокна).
Нормальное питание обусловлено несколькими потоками как нутритивных, так и регуляторных веществ, имеющих жизненно важное значение (гормоны и пептиды пищеварительной системы, вторичный поток вновь синтезируемых кишечной микрофлорой нутриентов и токсинов, резорбция продуктов незавершенного гидролиза пищи, а также различных токсинов, содержащихся в последней).
В трофическом отношении организм является надорганизменной структурой с своей собственной микрофлорой кишечника
11.