

II. Гипер- р-липопротеидемия делится на 2 типа:
Па - увеличение содержания в крови Р-ЛП при нормальном уровне пре-р-ЛП;
Пб - увеличение содержания Р-ЛП и пре-Р-ЛП.
Для заболевания характерен выраженный ксантоматоз век, кожи, роговицы, развитие ишемической болезни сердца с инфарктом миокарда в очень раннем возрасте, атеросклероти-ческие поражения сосудов у детей. Предполагается, что в основе заболевания лежит аутосом-но-доминантный дефект рецепторов ЛПНП (Па), либо нарушение взаимодействия рецепторов на клеточных мембранах с ЛПОНП и ЛПНП, либо дефект липопротеидлипазы (Пб).
III. «Флотирующая» гиперлипопротеиде-мия, или дис-р-липопротеидемия. В основе заболевания лежит наследственно обусловленное нарушение синтеза апопротеина Е (белок, входящий в состав ХМ и ЛПОНП). Заболевание характеризуется появлением в сыворотке фло-
тирующих р-ЛП, которые называются промежуточными. Они обогащены холестерином, а содержание триглицеридов в них может быть снижено. Образуются эти частицы при нарушении катаболизма ЛПОНП и ХМ. Встречаются также приобретенные формы заболевания при гипотиреозе, танжерской болезни, некоторых аутоиммунных гаммапатиях.
Этот вид ГЛП сопровождается ранними ате-росклеротическими проявлениями (после 20 лет), развитием ИБС, ишемической энцефалопатии вплоть до инсультов, ксантоматозом, ожирением.
IV. Гипер-пре-р-липопротеидемия. Заболевание может быть наследственно обусловленным (аутосомно-доминантное) или приобретенным (при алкоголизме, остром гепатите, акромегалии, диабете и др.). Патогенез до конца не выяснен. Для этого типа ГЛП характерно нарастание уровня триглицеридов и ЛПОНП в крови. Содержание ЛПНП и ЛПВП варьирует от нормального до значительно сниженного. У больных развиваются ожирение и сахарный диабет, появляются ксантомы, возможны атеросклеротическое поражение сосудов нижних конечностей, липи-доз сетчатки и ухудшение зрения, проявления ишемической болезни сердца.
V. Гипер-пре-р-липопротеидемия и хиломикронемия. При этом заболевании в крови увеличивается содержание ХМ и ЛПОНП и снижается уровень ЛПНП и ЛПВП. У больных отме-чаются гепато- и спленомегалия, ожирение, снижение толерантности к глюкозе, поражение миокарда. После приема жирной пищи могут наблюдаться внезапные приступы абдоминальной колики, ксантоматоз и атеросклероз слабо выражены.
В патогенезе первичного заболевания главную роль играет наследственно обусловленное отсутствие кофактора липопротеидлипазы - апопро-теина СП (аутосомно-рецессивное наследование), в результате два основных субстрата воздействия этого фермента накапливаются в крови.
Фенокопия болезни развивается при алкоголизме, гликогенозе Гирке и некоторых других заболеваниях печени.
Гипо-(а)липопротеидемии (относительно редкие аномалии спектра ЛП)
1. А-р-липопротеидемия. В основе заболевания лежит аутосомно-доминантный дефект синтеза апопротеина В (белковой части липопроте-идов), что приводит к аномалии строения хило-микронов, снижению содержания или полному отсутствию в плазме ЛПОНП и ЛПНП. Клинические проявления связаны с нарушением всасывания в кишечнике жиров и углеводов, гемолитической анемией, дегенерацией бокового и заднего канатиков спинного мозга, пигментной ретинопатией. Нарушение всасывания жиров проявляется сразу после рождения плохим аппетитом, рвотой, обильными испражнениями, стеатореей, развитием гипотрофии. Примерно у трети больных развивается умственная отсталость. С возрастом усиливаются неврологические расстройства, появляются скелетные деформации, сердечные аритмии, ухудшается зрение. В патогенезе заболевания решающее значение имеет снижение содержания холестерина в клеточных мембранах и потеря жирорастворимых витаминов, особенно витамина Е, что ведет к утрате антиоксидантной защиты мембран.
2. Танжерская (тэнжирская) болезнь. В основе заболевания лежит аутосомно-рецессивное нарушение синтеза апопротеина А, что, в свою очередь, нарушает продукцию ЛПВП. У больных нарушен транспорт эфиров холестерина, в результате эфиры захватываются макрофагами и откладываются в клетках ретикуло-эндотели-альной системы селезенки, печени, лимфоидных
органов. Наблюдается лимфаденопатия, гепато-спленомегалия, неврологические нарушения -слабость, парестезии, снижение сухожильных рефлексов. Одним из ярких признаков заболевания является оранжево-желтый цвет увеличенных миндалин.
Существуют и другие формы гиполипопроте-идемий: церебросухожильный ксантоматоз (наследственный дефект синтеза желчных кислот из холестерина), болезнь Вальмана (аутосомно-рецессивный дефицит холинэстеразы), гипоаль-фалипопротеинемия (генетически детерминированное нарушение продукции апопротеина А и С) и др. Большинство из них связано с наследственной патологией синтеза белковой части липопротеидов либо с нарушением метаболизма холестерина.
11.5.4. Нарушение депонирования жиров (ожирение и жировая инфильтрация печени)
Ожирение - избыточное отложение жира в жировой ткани. Считается, что человек страдает ожирением, если его масса превышает нормальную более чем на 20% и продолжает увеличиваться далее. Этим недугом страдает более трети взрослого населения России. В 1998 г. ВОЗ признала ожирение хроническим заболеванием. За последнее десятилетие число таких больных в мире увеличилось почти в два раза и по оценке специалистов в 2025 г. их количество составит 300 млн человек. Ситуация тем более сложная, что с каждым годом увеличивается число молодых людей, страдающих ожирением, снижается общая продолжительность жизни населения земного шара в связи с тяжелыми заболеваниями, сопутствующими ожирению (сахарный диабет, гипертония, атеросклероз) (табл. 42).
У тучных людей старше 50 лет смертность увеличивается на 50% по сравнению с лицами, не имеющими ожирения.
Классификация ожирения
Ожирение может возникать как самостоятельное заболевание - в этом случае говорят о первичном ожирении. Вторичное ожирение - это синдром, возникающий вследствие гормональных или других расстройств в организме.
Первичное ожирение возникает при нарушении гормональной связи между жировой тканьюи гипоталамусом. Его главная черта - абсолютная или относительная лептиновая недостаточность. В 1994 г. был обнаружен пептидный гормон адипоцитов (клетки жировой ткани) лептин. Клетки генерируют его при накоплении жира. Лептин действует на вентролатеральные ядра гипоталамуса и вызывает чувство сытости, вен-тромедиальные ядра (центр голода) при этом тормозятся. Существует тесная связь между продукцией лептина и выработкой гормонов желудочно-кишечным трактом, нейромедиаторов, инсулина, глюкокортикоидов и др. (рис. 93).
Вторичное ожирение - синдром, возникающий при нарушении соотношения между процессами липолиза и липогенеза, носит симптоматический характер и порождается различными эндокринными расстройствами (гиперкорти-золизм, гиперинсулинизм, гипогонадизм, опухоли мозга, нарушения мозгового кровообращения и пр.).
По степени увеличения массы тела различают ожирение I степени (масса тела увеличена на 30%); II степени (на 30-50%) и III степени (более чем на 50%).
Одним из наиболее распространенных показателей для оценки степени ожирения является индекс массы тела (ИМТ), рассчитываемый следующим образом:

Больные с ИМТ 30 кг/м2 и более, а также пациенты с ИМТ 27 кг/м2 или более, ожирение которых связано с такими факторами риска, как диабет II типа или дислипидемия, подлежат обязательному лечению (табл. 43).
Наиболее простым методом определения необходимости регулирования массы тела является измерение окружности талии. В идеале окружность талии не должна превышать 94 см у мужчин и 80 см у женщин. Если окружность талии у мужчин достигает 102 см, а у женщин -88 см, возникает серьезная угроза увеличения риска заболевания.
По особенностям морфологии жировой ткани выделяют гипертрофическое и гиперпластическое ожирение.
Гипертрофическое ожирение связано с увеличением размеров адипоцитов (это лабильный фактор, зависимый от питания), чаще встречается в зрелом возрасте. При этом виде ожирения масса тела может увеличиваться в 3-3,3 раза.
Гиперпластическое ожирение сопровождается увеличением количества адипоцитов. Начинается, как правило, в детском возрасте, так как дифференцировка фибробластических клеток-предшественниц в новые адипоциты во взрослом организме - явление довольно редкое (это происходит в период внутриутробного развития и в раннем грудном возрасте). Данный вид ожирения в большей степени зависит от наследственности. Избыток массы тела при гиперпластичес-

ком ожирении может достигать гигантских величин (до 1000%). Следует отметить, что в подростковый и преклимактерический периоды повышается пролиферативная активность преади-поцитов. Кроме того, их деление индуцируют избыточная калорийность пищи, сахарный диабет или переедание у беременных. В этих случаях гиперпластическое ожирение развивается у взрослых.
По патогенезу различают алиментарное, метаболическое и энергетическое ожирение.
Алиментарное ожирение - наблюдается при
чрезмерном потреблении пищи, что может быть обусловлено:
а) нарушением деятельности гипоталамичес-кого пищевого центра (длительное возбуждение вентролатеральных ядер) в результате травм, кровоизлияний в диэнцефальную область, воспаления (по этиологии это гипоталамическое ожирение);
б) частой афферентной импульсацией при возбуждении вкусовых рецепторов;
в) переходом от активного к малоподвижному образу жизни. При этом в некоторых случа-


ях сохраняется высокий уровень возбудимости пищевого центра (характерный для лиц физического труда или спортсменов), что приводит к систематическому перееданию;
г) чрезмерным растяжением стенок желудка при его переполнении. Это снижает чувствительность нервных окончаний слизистой оболочки, и тормозящие импульсы передаются в пищевой центр только при очень большом скоплении пищи в желудке. В результате переедание становится постоянным и возникает ожирение;
д) пожилым возрастом, что объясняется несоответствием между прежним уровнем возбудимости центра голода и меньшими энергозатратами (после 25 лет основной обмен снижается в каждые последующие 10 лет примерно на 7,5%). Интересно отметить, что в глубокой старости часто развивается исхудание, поскольку угнетается активность пищевого центра и снижается переход углеводов в жиры.
Метаболическое ожирение обусловлено повышенным синтезом жира из углеводов. В обычных условиях до 30% поступающей в организм глюкозы под действием инсулина превращается в жир. При гиперфункции инсулярного аппарата этот процент возрастает. Аналогичное изменение метаболизма наблюдается при повышенной продукции пролактина (гормона передней доли гипофиза), глюкокортикоидов.
Энергетическое ожирение развивается при недостаточном использовании жиров в качестве источника энергии. Наблюдается при гиподинамии в сочетании с хорошим аппетитом, при снижении тонуса симпатической нервной системы и недостаточной продукции жиромобилизующих гормонов (СТГ, тиреоидные гормоны, катехола-мины), поскольку задерживается выход жира из депо и использование его в качестве энергетического субстрата.
По этиологии ожирение классифицируют на экзогенно-конституциональное, гипоталамичес-кое, гормональное (эндокринное).
Экзогенно-конституциональное ожирение. Определенные условия способствуют реализации наследственно обусловленной предрасположенности. Длительное повышение активности «пищевого центра» ведет к повышению аппетита (гиперфагии) и ожирению (рис. 94). Привычка переедать может быть приобретена в детстве. Так, установлено, что избыточное кормление ребенка первого года жизни способствует развитию
гиперпластического ожирения, характеризующегося увеличением объема жировых клеток. В результате создаются условия для развития ожирения.
Гипоталамическое ожирение. Является следствием поражения области гипоталамуса. Причиной могут быть перенесенные травмы головного мозга, стойкая внутричерепная гипертен-зия, опухоли мозга, менингит, а также врожденные дегенеративные изменения гипоталами-ческой области (например, синдром Фрёлиха) (рис. 95).
Гормональное ожирение. Связано как с гипо, так и с гиперфункцией желез внутренней секреции и наблюдается при гипотиреозе, гипофункции половых желез, а также при гиперинсули-низме и гиперкортицизме. В крови таких больных повышается содержание ЛПНП и ЛПОНП, НЭЖК. При гормональном ожирении рано развивается гипертриацилглицеридемия и несколько позже - гиперхолестеринемия.
Нарушению обмена липидов сопутствует изменение углеводного обмена: развивается гипергликемия, стимулирующая секрецию инсулина и предшественника инсулина - низкоактивного гормона проинсулина. В свою очередь, секрецию проинсулина и инсулина стимулируют НЭЖК, ЛПНОП, ЛПНП. Усиленный выброс глюкокортикоидов, стимулирующих глюконеогенез, также повышает уровень инсулярных гормонов вкрови. Гиперинсулинизм ведет к усилению синтеза и депонирования липидов. Постепенно длительная стимуляция р-клеток инсулярного аппарата поджелудочной железы приводит к его истощению и провоцирует развитие сахарного диабета.
Последствия ожирения. При ожирении постепенно изменяется белковый обмен, который характеризуется снижением уровня общего белка крови преимущественно за счет уменьшения концентрации альбуминов, увеличением содержания фибриногена, продуктов деградации фибрина, снижением содержания гепарина. Следствием этого является нарушение транспорта НЭЖК и других липидов, повышение тромбо-генных свойств крови, снижение фибринолити-ческой активности крови, возникновение тром-боэмболических осложнений. Эти изменения являются факторами риска атеросклероза, ише-мической болезни сердца, инсульта, гипертони-
ческой болезни. Кроме этого, возникают изменения функций ЦНС, наблюдаются утомляемость, сонливость, ухудшение памяти, развивается преждевременное старение, возникают изменения во внутренних органах, например жировая инфильтрация (ожирение или жировая трансформация) печени (рис. 96).
Жировая инфильтрация печени. При нарушении расщепления и выведения жиров из клетки (гепатоцита) говорят о жировом пропитывании ее (инфильтрации). Сочетание инфильтрации с нарушением цитоплазматической структуры клетки и ее белковых компонентов называется жировой дистрофией (рис. 97).
Причинами жировой инфильтрации печени являются: сахарный диабет; ожирение; гипер-липопротеидемия; алкоголизм; отравление фосфором, мышьяком, хлороформом и другими ядами гепатотропного действия; голодание и гипо-витаминозы; инфекции и интоксикации; длительное стрессорное воздействие; недостаток в пище холина, метионина и других липотропных факторов, снижение синтеза липокаина в мелких протоках поджелудочной железы; беременность; наследственные дефекты окисления жирных кислот.
Патогенез ожирения печени связан с увеличением поступления липидов в гепатоциты и снижением их утилизации в результате торможения окисления свободных жирных кислот, нарушением образования ЛПОНП и их секреции в кровь. Например, гепатотропные яды уг-


нетают окисление свободных жирных кислот (СЖК) в митохондриях печени, нарушают образование ЛПОНП, образуют активные кислородные радикалы, повреждающие гепатоциты.
Ожирение печени может заканчиваться гибелью гепатоцитов и формированием фиброза и цирроза органа. В то же время следует знать, что в принципе это процесс обратимый и в некоторых случаях он может протекать бессимптомно. Однако чаще при ожирении печени выявляются патологические печеночные пробы, гипер-кетонемия и ацидоз (в крови обнаруживаются ацетон, ацетоуксусная и Р-оксимасляная кислоты), появляются признаки печеночно-клеточной недостаточности и энцефалопатии.
11.5.5. Нарушение обмена липидов и ненасыщенных жирных кислот
Жирные кислоты окисляются в процессе (3-окисления, образуя ацетил-КоА. В печени из
двух молекул ацетил-КоА образуется ацетоаце-тил-КоА, который затем взаимодействует еще с одной молекулой ацетил-КоА. Образовавшийся (3-окси-Р-метилглутарил-КоА способен под действием фермента оксиметилглутарил-КоА-лиа-зы расщепляться на ацетоацетат и ацетил-КоА. При декарбоксилировании ацетоуксусной кислоты (ацетоацетат) образуется ацетон, при ее восстановлении - D-P-оксимасляная кислота (D-Р-оксибутират). Существует второй путь синтеза кетоновых тел: при отщеплении КоА от аце-тоацетил-КоА образуется свободная ацетоуксусная кислота. Процесс катализируется ферментом деацилазой, однако его активность в печени низкая, следовательно, этот путь не имеет существенного значения. При жировой инфильтрации печени отмечается избыточное поступление НЭЖК, задержка их окисления и выведения липопротеидов из печени, происходит накопление кетоновых тел в крови (гиперкетоне-мия) и в моче (кетонурия). Возможно изменение рН крови.
Гиперкетонемия со сдвигом рН крови в кислую сторону (ацидоз) возможна при угнетении цикла Кребса (цикла трикарбоновых кислот), в котором происходит окисление кетоновых тел. Это наблюдается при голодании, сахарном диабете, стрессе различной этиологии, тяжелой мышечной работе. Однако следует помнить, что в экстремальных ситуациях из кетоновых тел путем глюконеогенеза может синтезироваться глюкоза, служащая источником энергии для работы центральной нервной системы.
При метаболизме липидов образуются полиненасыщенные высшие жирные кислоты (лино-левая, линоленовая, арахидоновая). Они являются регуляторами жизненно важных функций и процессов гомеостаза: контролируют течение иммунных реакций, физиологических родов, репаративных процессов. Во время метаболизма арахидоновой кислоты по циклооксигеназ-ному пути в организме образуются простаглан-дины, простациклины, тромбоксаны, а при работе липоксигеназного метаболического пути из арахидоновой кислоты образуются лейкотриены.
Простагландины (ПГ) G2 и Н2 являются предшественниками стабильных ПГЕ, ПГ , ПГГ а также простациклинов и тромбоксанов. ПГЕ оказывают иммуномодулирующее действие, расслабляют гладкие мышцы бронхов, трахеи, ЖКТ, тормозят высвобождение медиаторов из тучныхклеток. ПГВ2 сокращают гладкую мускулатуру бронхов и желудочно-кишечного тракта, оказывают вазодилататорное действие, снижая артериальное давление. ПГр сокращают гладкую мускулатуру, повышают проницаемость сосудов, стимулируют высвобождение медиаторов из тучных клеток.
Тромбоксаны синтезируются преимущественно в ткани мозга, селезенки, легких и в тромбоцитах, клетках воспалительной гранулемы. Они вызывают адгезию и агрегацию тромбоцитов, способствуют развитию тромбоза при ишемичес-кой болезни сердца, оказывают вазоспастичес-кое действие.
Простациклины (например, ПГЛ2) синтезируются в эндотелии сосудов, сердце, ткани матки, слизистой желудка, расслабляют гладкую мускулатуру сосудов, вызывают дезагрегацию тромбоцитов, способствуют фибринолизу.
Лейкотриены (А, В, С, D) синтезируются во всех клетках крови (кроме эритроцитов), а также в адвентиции сосудов, в тучных клетках, легких. Они способствуют сокращению гладкой мускулатуры дыхательных путей и желудочно-кишечного тракта, оказывают сосудосуживающее действие (в том числе коронарных артерий). Лейкотриены запускают синтез простагландинов и простациклинов при угнетении выброса тром-боксана, что наблюдается при анафилактическом шоке. Органом-мишенью для лейкотриенов является сердце. Выделяясь в избытке, они ин-гибируют (на 60%) сократимость сердечной мышцы, уменьшая коронарный кровоток, усиливая воспалительную реакцию.
Таким образом, биологически активные вещества, являющиеся продуктами полиненасыщенных жирных кислот, в оптимальных условиях сохраняют гомеостаз организма. В случае изменения их количественного соотношения могут развиваться различные патологические реакции.
11.5.6. Нарушение обмена фосфолипидов
Описаны некоторые наследственные заболевания и патологические состояния, связанные с избыточным отложением в тканях фосфолипидов.
Так, при болезни Гоше цереброизиды откладываются в макрофагальных клетках селезенки, печени, лимфатических узлах и в костном
мозге. При болезни Нимана - Пика в клетках различных органов наблюдается отложение фос-фатида сфингомиелина. При амавротической (от греч. amauros - темный, слепой) семейной идиотии липиды отлагаются в нервных клетках, что сопровождается атрофией зрительных нервов и слабоумием.
11.5.7. Нарушение обмена холестерина
Холестерин (ХН) - производное циклопен-тана и гидратированного фенантрена. Его название происходит от греческих слов «желчь» и «твердый», поскольку впервые описан в XVIII столетии как компонент желчных камней. Общее содержание ХН в организме человека от 100 до 300 г, преобладает свободный ХН (его почти в 3 раза больше, чем эфиров холестерина). ХН может поступать в организм с пищей (яичный желток, печень, мясо, сливочное масло, сметана и сливки), существует также эндогенный синтез холестерина в печени из ацетил-КоА. Кроме того, печень - это единственный орган, где образуются эфиры ХН, поэтому снижение уровня этери-фицированного ХН служит одним из показателей недостаточности функции печени. Суточное потребление ХН варьирует от 0,2 до 0,5 г, в самом организме образуется около 1 г в день. Роль ХН огромна, он входит в состав клеточных мембран, изменяя их жидкие свойства и проницаемость, влияет на активность мембранных ферментов, может стимулировать пролиферацию способных к делению клеток (при его избытке в мембране). Холестерин является предшественником желчных кислот и стероидных гормонов: половых, глюкокортикоидов, минералокортико-идов, а также витамина D.
Выведение холестерина из организма осуществляется несколькими путями - около 0,5 г ХН в сутки превращается в желчные кислоты и теряется с желчью через кишечник, примерно такое же его количество теряется ежесуточно с фекалиями (копростерин), наконец, около 0,1 г ХН выделяют сальные железы.
В плазме крови здорового человека содержится 5,2 - 6,2 ммоль/л холестерина. Патогенными для организма являются как избыток, так и недостаток ХН.
К возникновению гиперхолестеринемии могут привести:
1. Возбуждение симпатической нервной сие-темы (стресс), что способствует усиленной мобилизации жира из депо и синтезу эндогенного ХН.
2. Нарушение ресинтеза жирных кислот из ацетил-КоА, что наблюдается при сахарном диабете.
3. Нарушение выведения ХН из организма при угнетении перистальтики кишечника, дис-кинезии желчных путей, при механической желтухе.
4. Эндокринные заболевания, нарушающие обмен липидов: гипотиреоз, гиперкортицизм.
5. Беременность.
6. Нефротический синдром (нарушения ли-пидного обмена и снижение содержания альбуминов).
7. Гиповитаминоз С, гипоксии, поскольку распад ХН требует достаточного количества АТФ.
8. Повышенное поступление с пищей (однако в этом случае угнетается синтез эндогенного ХН).
9. При усиленном поступлении в организм животных жиров и рафинированных углеводов повышается синтез эндогенного ХН.
10. Наследственно обусловленные дефекты ферментов обмена липидов (в том числе ХН).
К последствиям гиперхолестеринемии следует отнести развитие атеросклероза, ксантома-тоза, холестеатоза (отложение ХН и его эфиров в паренхиматозных органах с последующим развитием цирроза), ожирения, ИБС, рассеянного склероза (наследственные формы накопления ХН), ретинопатии и др.
Среди причин гипохолестеринемии следует указать на:
1. Наследственно обусловленные абета- и анальфалипопротеинемиями.
2. Заболевания печени с утратой способности к синтезу ХН и его эфиров.
3. Гипертиреоз.
4. Неполное голодание (сниженное поступление продуктов, богатых холестерином, животных жиров, рафинированных углеводов).
5. Некоторые виды анемий.
6. Усиление выведения ХН при поносах.
Последствиями гипохолестеринемии являются: 1) нарушение барьерной функции клеточных мембран, повышение ее проницаемости и цитолиз (также гемолиз эритроцитов); 2) неврологические нарушения, связанные с нарушением структуры миелиновых нервных волокон и проведения нервного импульса (атаксия, гипо-рефлексия, парестезии); 3) снижение образования желчных кислот и, следовательно, наруше-

ние пищеварения в кишечнике (потеря жиров и жирорастворимых витаминов); 4) гиповитаминоз D и соответствующие ему изменения; 5) гипо-продукция стероидных гормонов.
Нарушение регуляции скорости биосинтеза холестерина. В клетке липопротеиды расщепляются под влиянием ферментов лизосом с освобождением неэтерифицированного холестерина (рис. 98). По мере накопления холестерина в клетке ограничивается скорость его образования, угнетается активность фермента гидроксиметил-глютарил-КоА (ГМГ-КоА-редуктазы), который является ключевым ферментом, регулирующим скорость биосинтеза холестерина. Этот процесс возможен во всех клетках, кроме эритроцитов. Этот контроль предупреждает развитие атеросклероза. При недостаточности ЛПНП-рецепторов в тканях повышается уровень ЛПНП в крови, продлевается время их циркуляции, что способствует образованию модифицированных формЛПНП, обладающих высокой атерогенностью при неспецифическом эндоцитозе (избыточном неконтролируемом поступлении модифицированных ЛПНП в сосудистую стенку). Развивается атеросклероз.
Таким образом, бесконтрольное поступление липопротеидов в стенку сосудов, освобождение холестерина из молекулы ЛПНП и нарушение выведения его из клетки сосудистой мембраны, а также задержка утилизации холестерина в печени ведут к возникновению атеросклероза и ишемической болезни сердца (см. гл.14).
11.6. ПАТОФИЗИОЛОГИЯ БЕЛКОВОГО ОБМЕНА
Белки, наряду с нуклеиновыми кислотами, при участии которых они образуются, являются основой всех жизненных процессов. Это важнейший структурный материал, необходимый для построения клеток и межклеточного вещества; белки входят как обязательная часть в состав всех ферментов, управляющих обменными процессами; значительная часть гормонов также являются белками (полипептидами). Белки играют важную роль в транспорте с током крови липидов, жирных кислот, гормонов, витаминов, неконъюгированного билирубина, микроэлементов и других веществ; при их участии осуществляются свертывание крови, иммунные реакции, поддержание кислотно-щелочного равновесия; они создают онкотическое давление плазмы крови, что играет важную роль в осуществлении процессов микроциркуляции и водного обмена. Белки являются источником энергии - при полном распаде и окислении 1 г белка освобождается 4,1-4,3 ккал тепла, однако эта функция белков легко возмещается окислением углеводов и жиров.
Нарушение обмена белков может происходить на различных этапах: при расщеплении и усвоении белков пищи, их внутриклеточном синтезе и распаде, образовании конечных продуктов и их экскреции.
11.6.1. Нарушение расщепления пищевых белков и усвоения образующихся аминокислот
В желудке и кишечнике происходит гидролитическое расщепление белков пищи до пепти-
дов и аминокислот под влиянием ферментов желудочного сока (пепсин, гастриксин), панкреатического (трипсин, химотрипсин, эластаза и карбоксипептидазы) и кишечного (аминопеп-тидаза, дипептидазы) соков. Образующиеся при расщеплении белков аминокислоты всасываются стенкой тонкого кишечника в кровь и потребляются клетками различных органов. Нарушение этих процессов имеет место при заболеваниях желудка (воспалительные и язвенные процессы, опухоли), поджелудочной железы (панкреатиты, закупорка протоков, рак), тонкого кишечника (энтериты, диарея, атрофия слизистой, состояния мальдигестии и мальабсорбции). Такие обширные оперативные вмешательства, как удаление желудка или значительной части тонкого кишечника, также сопровождаются нарушением расщепления и усвоения белков пищи. Усвоение пищевых белков нарушается при лихорадке вследствие снижения секреции пищеварительных ферментов.
Недостаточность усвоения белков пищи сопровождается дефицитом аминокислот и нарушением синтеза собственных белков. Недостаток пищевых белков не может быть полностью компенсирован избыточным введением и усвоением каких-либо других веществ, так как белки являются основным источником азота для организма.
11.6.2. Нарушение процессов эндогенного синтеза белка
Синтез белков происходит в организме непрерывно на протяжении всей жизни, но наиболее интенсивно совершается в период внутриутробного развития, в детском и юношеском возрасте.
Различают следующие виды синтеза белка в зависимости от его назначения:
- регенерационный, связанный с процессами физиологической и репаративной регенерации;
- синтез роста, сопровождающийся увеличением массы и размеров тела;
- стабилизирующий, связанный с возмещением структурных белков, утраченных в процессе диссимиляции, способствующий поддержанию структурной целостности организма;
- функциональный, связанный со специфической деятельностью различных органов (синтез гемоглобина, белков плазмы крови, антител, гормонов и ферментов).

Причинами нарушения синтеза белка в организме могут являться отсутствие или недостаток одного или более факторов, участвующих в этом процессе:
- отсутствие достаточного количества аминокислот;
- дефицит энергии в клетках;
- расстройства нейроэндокринной регуляции;
- нарушение процессов транскрипции или трансляции информации о структуре того или иного белка, закодированной в геноме клетки.
Наиболее частой причиной общего нарушения эндогенного синтеза белка является недостаток аминокислот в организме вследствие: 1) расстройств пищеварения и всасывания; 2) пониженного содержания белка в пище; 3) питания неполноценными белками, в составе которых отсутствуют или имеются лишь в незначительном количестве незаменимые аминокислоты, не синтезирующиеся в организме.
К числу незаменимых относятся такие аминокислоты, как валин, лизин, триптофан, лейцин, изолейцин, треонин, метионин, фенилала-нин и гистидин (табл. 44). Полный набор незаменимых аминокислот имеется в большинстве белков животного происхождения, тогда как растительные белки могут не содержать некоторые из них или содержат в незначительном количестве (например, в белках кукурузы мало триптофана). Недостаток в организме хотя бы одной из незаменимых аминокислот ведет к снижению эндогенного синтеза того или иного белка даже при изобилии остальных.
Дефицит в пище заменимых аминокислот значительно реже приводит к понижению син-
теза белка, так как они могут образовываться в организме из кетокислот, являющихся продуктами метаболизма углеводов, жиров и белков. Недостаток кетокислот может возникнуть при сахарном диабете, при нарушении процессов окислительного дезаминирования и трансамини-рования аминокислот (гиповитаминоз В6).
Синтез белка является энергозависимым процессом. Энергия, освобождающаяся при расщеплении макроэргических соединений АТФ и ГТФ, необходима для активации аминокислот и для образования пептидных связей (в количестве 21,9 кал на каждую пептидную связь). Недостаток в клетках источников энергии имеет место при гипоксии, действии разобщающих факторов, сахарном диабете, гиповитаминозе Bj, дефиците никотиновой кислоты и др.
Роль нервной и эндокринной систем в регуляции процессов синтеза и расщепления белка
Нервная система оказывает на белковый обмен прямое и косвенное действие. Подтверждением прямого трофического влияния нервной системы на метаболизм белков в клетках является развитие атрофических и дистрофических изменений в денервированных тканях. Установлено, что в денервированных тканях процесс распада белка превалирует над синтезом. Косвенное влияние нервной системы на белковый обмен осуществляется путем изменения функции эндокринных желез.
Действие гормонов может быть анаболическим (усиливающим синтез белка) и катаболи-ческим (повышающим распад белка в тканях). Выраженное анаболическое действие на общий синтез белка оказывают инсулин, соматотроп-ный гормон и тиреоидные гормоны в физиологических дозах. Половые гормоны оказывают анаболический эффект на синтез белка в половых органах; тестостерон, кроме того, стимулирует синтез белка в мышцах, а эстрадиол - в молочных железах, костях и хрящах, способствуя росту.
Катаболическое действие оказывают тиреоидные гормоны при повышенной их продукции (гипертиреоз). Глюкокортикоиды усиливают синтез белков и нуклеиновых кислот в печени и повышают распад белков в других органах и тканях, включая мышцы, кожу, кости, лимфо-идную и жировую ткани; повышенное содержание глюкокортикоидов в организме усиливает гибель лимфоцитов и инволюцию лимфоидной ткани, подавляет иммунный ответ.
Анаболическое действие гормонов осуществляется путем активации определенных генов, в связи с чем усиливается образование различных видов РНК (информационная, транспортная, рибосомальная) и ускоряется синтез белков; механизм катаболического действия гормонов изучен недостаточно, но он связан с повышением активности тканевых протеиназ.
Снижение синтеза гормонов анаболического действия, таких как СТГ и тиреоидные гормоны, в детском возрасте ведет к задержке роста.
Инактивацию тех или иных факторов, участвующих в биосинтезе белка, могут вызвать некоторые лекарственные препараты (например, антибиотики) и микробные токсины. Известно, что дифтерийный токсин тормозит присоединение аминокислот к синтезируемой полипептидной цепи; этот эффект устраняется анатоксином.
Стимулирующее или угнетающее действие на синтез белка могут оказать изменения концентрации различных ионов (прежде всего Mg2+), уменьшение или увеличение ионной силы.
Последствия общего нарушения синтеза белка
Длительное и значительное понижение синтеза белка приводит к развитию дистрофических и атрофических нарушений в различных органах и тканях вследствие недостаточного обновления структурных белков. Замедляются процессы регенерации. В детском возрасте тормозятся рост и развитие. Снижается синтез различных ферментов и гормонов (СТГ, антидиуре-
тический и тиреоидный гормоны, инсулин и др.), что приводит к нарушению других видов обмена, в частности к снижению основного обмена. Понижается содержание белков в сыворотке крови в связи со снижением их синтеза в гепатоци-тах. Вследствие этого в крови уменьшается он-котическое давление, что способствует развитию отеков, так как при гипопротеинемии вода слабо удерживается в кровеносном русле. Уменьшается продукция антител и других защитных белков и, как следствие, снижается иммунологическая реактивность организма. В наиболее выраженной степени эти расстройства возникают в результате длительного нарушения усвоения белков пищи при различных хронических заболеваниях органов пищеварения, а также при длительном белковом голодании, особенно если оно сочетается с дефицитом жиров и углеводов. В последнем случае повышается использование белка в качестве источника энергии.
Причины и механизм нарушения синтеза отдельных белков
Такие нарушения в большинстве случаев имеют наследственную природу. В основе их лежит отсутствие в клетках информационной РНК (иРНК), специфической матрицы для синтеза какого-либо определенного белка, или нарушение ее структуры вследствие изменения структуры гена, на котором она синтезируется. Генетические нарушения, например замена или потеря одного нуклеотида в структурном гене, приводят к синтезу измененного белка, нередко лишенного биологической активности.
К образованию аномальных белков могут привести не только нарушение процесса транскрипции (отклонения от нормы в структуре иРНК), но и дефекты трансляции, т.е. передачи информации относительно структуры синтезируемой на рибосомах полипептидной цепочки какого-либо белка. Одной из причин этого может быть мутация транспортной РНК, вследствие чего к ней присоединяется несоответствующая аминокислота, которая и будет включаться в полипептидную цепь при ее сборке (например, при образовании НЬ).
Процесс трансляции является сложным, совершающимся при участии ряда ферментов, и нарушение функции какого-либо из них может привести к тому, что та или другая иРНК не передаст закодированную в ней информацию.
Нарушение синтеза отдельных белков-фермен-тов или структурных белков лежит в основе различных наследственных болезней (гемоглобино-зы, альбинизм, фенилкетонурия, галактоземия, гемофилия и многие другие). Нарушение какой-либо ферментативной функции чаще всего связано не с отсутствием соответствующего белка -фермента, а с образованием патологически измененного неактивного продукта.
11.6.3. Причины, механизм и последствия повышенного распада тканевых белков
Наряду с синтезом в клетках организма постоянно происходит деградация белков под действием протеиназ. У здорового взрослого человека процессы распада и синтеза белка уравновешены, т.е. имеется азотистое равновесие. При этом суточная деградация белка составляет 30-40 г. Обновление белков за сутки у взрослого человека составляет 1-2% от общего количества белка в организме и связано преимущественно с деградацией мышечных белков [Марри Р. и со-авт., 1993], при этом 75-80% освободившихся аминокислот вновь используется для синтеза.
У детей и беременных женщин синтез белка преобладает над его распадом, вследствие этого количество азота, поступающего в организм, превалирует над общим количеством экскрети-руемого азота (положительный азотистый баланс). При патологии распад белка может превалировать над синтезом и азота поступает в организм меньше, чем выделяется (отрицательный азотистый баланс). Превалирование процессов деградации белка над синтезом наблюдается при инфекционной лихорадке, обширных травмах, ожогах и воспалительных процессах, прогрессирующем злокачественном опухолевом росте, некоторых эндокринных заболеваниях (сахарный диабет, гипертиреоз), при тяжелом эмоциональном стрессе, обезвоживании, белковом голодании, лучевой болезни. В механизме усиленного распада белков при многих из перечисленных состояний лежит повышенная продукция катаболических гормонов.
Азотистое равновесие нарушается (отрицательный азотистый баланс) при гиповитамино-зах А, С, Bj, В2, В6, РР при дефиците фолиевой кислоты. Следствием отрицательного азотистого баланса являются дистрофические изменения в органах, похудание, в детском возрасте - задержка роста.
11.6.4. Нарушение обмена аминокислот
Аминокислоты поступают в кровь и ткани из пищеварительного тракта; кроме того, они образуются при деструкции тканевых белков под действием внутриклеточных катепсинов (протеиназ).
Основная часть аминокислот используется в организме в качестве строительных блоков при синтезе белков. Кроме того, аминокислоты используются для синтеза пуриновых и пирими-диновых оснований, гормонов, тема, различных биологически активных пептидов (интерлейки-ны, факторы роста и т.д.), меланина, глюкозы, жирных кислот и ряда других веществ. Глицин и глутамат играют роль нейромедиаторов в ЦНС. Аминокислоты, не использованные для вышеупомянутых целей, подвергаются окислению до С02 и Н20 с освобождением энергии. В норме при окислении аминокислот освобождается 10-15% образующейся в организме энергии. Окисление аминокислот усиливается при избыточном поступлении их в организм, при голодании, сахарном диабете, гипертиреозе, снижении синтеза белков и некоторых других состояниях.
Окислению аминокислот предшествует отщепление от них аминогруппы и превращение в а-кетокислоты. Согласно существующим представлениям дезаминирование аминокислот осуществляется в два этапа. Первоначально происходит перенос аминогруппы аминокислоты на а-кетоглутаровую кислоту (трансаминирова-ние). В результате образуются глутаминовая кислота и та или иная кетокислота (например, из аланина - пировиноградная).

Процесс трансаминирования катализируется трансаминазами, коферментом которых является пиридоксальфосфат. Образовавшаяся при этом процессе глутаминовая кислота подвергается окислительному дезаминированию, т.е. отщеплению аминогруппы под действием глутаматде-гидрогеназы с образованием иона аммония (NH4+) и ct-кетоглутаровой кислоты, которая может снова вступить в реакцию трансаминирования или окислиться в цикле трикарбоновых кислот. Ке-токислоты, образующиеся при трансаминирова-нии (например, пировиноградная), также могут окислиться до С02 и Н20 подобно глюкозе и жирным кислотам. Поскольку реакции трансаминирования и окислительного дезаминирова-ния могут идти как в прямом, так и в обратном направлении, то они играют роль не только в превращении аминокислот в кетокислоты, но и в образовании из кетокислот ряда заменимых аминокислот в том случае, если организм испытывает в них потребность. Кроме того, кетокислоты могут быть использованы для синтеза глюкозы.
Нарушение процесса трансаминирования в целом организме происходит при гиповитаминозе В6, при недостатке сс-кетокислот (голодание, сахарный диабет). Нарушение трансаминирования в отдельных органах, например в печени, происходит при некрозе клеток, что сопровождается выходом трансаминаз в кровь. Такое же явление имеет место при инфаркте миокарда. В поврежденных клетках может быть нарушен синтез белковой части трансаминаз.
Процесс окислительного дезаминирования снижается не только в связи с ослаблением трансаминирования, но и при гипоксии, гипо-витаминозах В2, РР, С, белковом голодании.
Нарушение процессов трансаминирования и окислительного дезаминирования аминокислот ограничивает их использование для синтеза глюкозы, жирных кислот, заменимых аминокислот, а также их окисление с освобождением энергии. При этом повышается содержание свободных аминокислот в сыворотке крови и в моче (ги-пераминоацидемия и гипераминоацидурия), снижается синтез мочевины. Такие нарушения особенно выражены при обширных повреждениях гепатоцитов (вирусные и токсические гепатиты и др.), так как в этих клетках метаболизм аминокислот происходит наиболее интенсивно.
Наряду с вышеупомянутой внепочечной ги-пераминоацидурией, обусловленной усиленным
поступлением аминокислот из крови в мочу, существует почечная форма гипераминоациду-
рии, связанная с нарушением реабсорбции аминокислот в почечных канальцах, при этом содержание аминокислот в сыворотке крови нормально или даже понижено (см. гл. 18). Гипераминоацидурия (физиологическая) может наблюдаться у детей раннего возраста в связи с функциональной неполноценностью (незрелостью) эпителия почечных канальцев; у беременных женщин повышается экскреция с мочой гисти-дина и ряда других аминокислот.
Одним из путей метаболизма аминокислот является их декарбоксилирование, которое состоит в отщеплении от аминокислоты С02. В результате образуются биогенные амины: гиста-мин - из гистидина, серотонин - из 5-окситрип-тофана, тирамин - из тирозина, у-аминомасля-ная кислота (ГАМК) - из глутаминовой, дофамин - из диоксифенилаланина и некоторые дру-
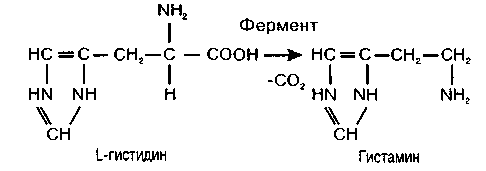
Этот процесс катализируется декарбоксила-зами, коферментом которых является пиридоксальфосфат (витамин Вв); при его дефиците образование биогенных аминов снижается. В частности, уменьшается образование у-аминомасля-ной кислоты, которая является основным тормозным нейромедиатором, как следствие этого наблюдается частое развитие судорог. Биогенные амины обладают высокой физиологической активностью. Наряду с ГАМК, серотонин и дофамин являются также нейромедиаторами в ЦНС, их повышенное или пониженное содержание в ткани мозга играет роль в патогенезе некоторых форм нейропатологии (нервной депрессии, паркинсонизма, шизофрении). Повышенное образование в организме серотонина, наиболее выраженное при карциноиде (опухоль, развивающаяся из энтерохромафинных клеток кишечника), сопровождается спазмом мускулатуры бронхов и кишечника, диареей, усилением агрегации тромбоцитов; кроме того, серотонин является мощным вазоконстриктором. Хорошо известна роль гистамина в появлении болевых ощущений, развитии воспаления и аллергических реакций, в том числе анафилактического шока.
Устранение избытка биогенных аминов происходит при участии аминооксидаз, которые катализируют превращение их в альдегиды после отщепления аминогруппы в виде NH3. Серо-тонин превращается в оксииндолилуксусную кислоту, которая выделяется с мочой.
Наследственные нарушения обмена некоторых аминокислот. Существуют многочисленные заболевания, обусловленные нарушением метаболизма аминокислот. С расстройствами метаболизма фенилаланина связано заболевание фе-нилкетонурией. К этому приводит мутация гена, необходимого для образования фермента фенил-аланингидроксилазы, при участии которой происходит превращение фенилаланина в тирозин. При отсутствии данного фермента наблюдается накопление в организме фенилаланина и промежуточных продуктов его метаболизма - фенил-пировиноградной, фенилуксусной и фенилмолоч-ной кислот, которые оказывают токсическое действие на мозг ребенка. Фенилпируват выделяется с мочой, где его можно обнаружить. Основные проявления фенилкетонурии - умственная отсталость, психозы, судорожные припадки, экзема, мышиный запах [Марри Р. и соавт., 1993]. Предотвратить развитие болезни можно только ранним переводом ребенка на диету с очень низким содержанием фенилаланина. Болезнь наследуется по аутосомно-рецессивному типу.
Одним из заболеваний, обусловленных нарушением метаболизма тирозина, является алкап-тонурия. Развитие ее связано с генетически обусловленным дефицитом фермента оксидазы гомо-гентизиновой кислоты, которая является одним из продуктов метаболизма тирозина. В связи с указанным дефектом гомогентизиновая кислота в большом количестве выделяется с мочой, придавая ей темно-коричневую окраску. Кроме того, гомогентизиновая кислота накапливается в соединительной и хрящевой тканях, также обусловливая их темное окрашивание (охроноз). Может развиться артрит. Передача дефектного гена осуществляется по аутосомно-рецессивному типу. Нарушением метаболизма тирозина обусловлены и такие заболевания, как тирози-ноз (тирозинемия) и альбинизм.
Гистидинемия - заболевание, связанное с замедлением превращения гистидина в уроканат вследствие дефицита фермента гистидазы. В
крови и моче обнаруживается повышенное содержание гистидина. Большинство больных ги-стидинемией характеризуются умственной отсталостью и дефектами речи. Заболевание наследуется по аутосомно-рецессивному типу.
Цистиноз - наследственное заболевание, характеризующееся отложением кристаллов цис-тина во многих тканях и органах, что связывают с нарушением функции лизосом. В моче повышено содержание всех аминокислот. Летальный исход наступает в раннем детском возрасте вследствие развития острой почечной недостаточности.
11.6.5. Нарушение конечного этапа обмена белка и аминокислот
Конечным продуктом обмена белка и аминокислот является мочевина, выделяющаяся из организма с мочой. Синтез мочевины осуществляется гепатоцитами в орнитиновом цикле. Образование мочевины имеет большое физиологическое значение, так как благодаря этому процессу происходит обезвреживание высоко токсичного продукта - аммиака, отщепляющегося от аминокислот при их дезаминировании, а также поступающего в кровь из кишечника. Обезвреживание аммиака, образующегося в клетках различных органов, в том числе в мозге, достигается путем реакции амидирования, т.е. присоединение его к аспарагиновой и в особенности глутаминовой кислотам с образованием аминов аспарагина и глутамина. Процесс амидирования, так же как и образование мочевины, идет с потреблением энергии, источником которой является АТФ.
Синтез мочевины понижается при длительном белковом голодании (недостаток ферментов), при заболеваниях печени (циррозы, острые гепатиты с повреждением большого числа гепато-цитов, отравление печеночными ядами), а также при наследственных дефектах синтеза ферментов, участвующих в орнитиновом цикле образования мочевины (карбамилфосфатсинтетазы, аргининсукцинатсинтетазы и аргининсукцинат-лиазы). При нарушении синтеза мочевины количество ее в крови и моче снижается и нарастает содержание аммиака и аминокислот, т.е. резидуального азота (продукционная гиперазотемия). Гипераммониемия играет важную роль в патогенезе печеночной энцефалопатии и комы.
Избыток аммиака может в некоторой степени устраняться за счет повышенного образования глутамина и присоединения к а-кетоглутаровой кислоте, которая при этом превращается в глу-таминовую, и ее окисление в цикле трикарбоно-вых кислот резко снижается. Вследствие этого снижается образование АТФ.
Другой причиной накопления небелковых азотистых продуктов в крови (креатинин, мочевина) является нарушение выделительной функции почек при острой и хронической почечной недостаточности или при нарушении проходимости мочевыводящих путей. Возникающая в данном случае гиперазотемия называется ретен-ционной. При этом концентрация остаточного азота в крови возрастает до 140-215 ммоль/л, а содержание небелковых азотистых продуктов в моче снижается. Ретенционная гиперазотемия является одним из факторов, играющих роль в развитии уремической комы.
Возможно развитие смешанной (комбинированной) формы гиперазотемии, при которой повышенный распад белка в тканях сочетается с недостаточным выведением азотистых продуктов с мочой. Такое сочетание возможно при острой почечной недостаточности, развившейся на почве септического аборта, или обширном сдав-лении тканей (синдром раздавливания). К комбинированной форме гиперазотемии относится гипохлоремическая гиперазотемия, возникающая при неукротимой рвоте, стенозе привратника и профузных поносах.