

1.ПРЕДМЕТ ОРГАН. ХИМИИ.Органич.химия-наука, изучающая соединения углерода.Атом углерода образует прочные связи не только с другими атомами, но и между собой, что определяет возможности возникновения линейных и циклических структур. Углерод 4-х валентен, что позволяет углер.цепям разветвляться, образуя сложные структуры. Углерод способен образовывать кратные связи с атомами углерода и другими элементами.Осн.направления развития органич.химии. Некоторые органические соединения были известны еще в глубокой древности: сахар (С12Н22О11), спирт (С2Н6О), уксусная кислота (С2Н4О2), растительные масла и животные жиры, индиго (С16Н10О2N2), лизарин (С14Н8О4), пурпур (С16Н8Br2O2N2), метиловый спирт (СН4О) и ацетон (С3Н6О).Я.Берцелиус разделил все вещества на 2 класса-органические и неорганические. Органические образуются только в живых организмах под действием «жизненной силы» (vis vitalis). Виталическая теория пользовалась большой популярностью, хотя появлялись факты ей противоречащие: синтез щавелевой кислоты, мочевины, ацетилена из карбида кальция.А.М.Бутлеров синтезировал сахаристое вещество – акрозу из формальдегида в 1881 г.М.Г. Кучеров синтезировал уксусную кислоту, Бертло – жиры. Теория радикалов:все органич.соединения состоят из 2-х частей: радикала и соединенных с ним групп. Теория типов: органич. вещества классифицируют по типам превращений: типы водорода, хлористого водорода, аммиака, воды и метана.Унитарная теория объединила теорию радикалов и теорию типов. Соединение-цельная система, составленная определенным, но неизменным порядком атомов, строение молекул можно отразить только реакциями, но не химическими формулами.Ф.А.Кекуле сформулировал понятие о 4-х валентности атома углерода и способности связываться друг с другом с образованием цепей. А.М.Бутлеров предположил, что для каждого соединения возможна только одна структурная формула, отражающая его свойства.Осн.положения теории:1.химические свойства определяются природой составных частиц, их количеством и строением.2.у веществ с одинаковым составом может быть различное строение – изомеры.3.в молекуле существует строгая последовательность хим.связывания атомов.4.реакционная способность атомов молекулы изменяется в зависимости от окружения.5.физ.и хим. свойства органич.соединений определяются составом молекул, химическим, пространственным и электронным строением. 6.строение вещества можно изучать хим. и физ. методами.
2.ЭЛЕКТРОН ТЕОРИЯ ХИМ СВЯЗИ. Хим.связь-любое взаимодействие между двумя или более частицами, сопровождающееся выделением энергии.Типы хим.связей:1.Ковалентные: 1)собственно-ковалентные: полярные, неполярные и сигма и пи 2)донорно-акцепторные:семиполярные 2.Электротстатические: 1) ионные 2) ион-дипольные 3)диполь-дипольные: водородные(внутримолекулярные и межмолекулярные) 4) ван-дер-ваальсовы. В органич.соединениях преобладает ковалентная связь, которая характеризуется:-длиной,-валентным углом,-энергией,-полярностью,-насыщаемостью,-направленностью в пространстве.Под длиной связи понимают равновесие между ядрами связанных атомов. Половина длины связи в симметричной молекуле называется ковалентным радиусом.И.Ленгмюр (1919) ввел понятие ковалентная связь, под которой понимается связь, образованная парой электронов. Общая электронная пара ковалентной связи может быть образована несколькими способами:- коолигация электронов;- координация электронов.В первом случае – каждая частица предоставляет по одному неспаренному электрону. Во втором- одна частица дает электронную пару, вторая предоставляет незаполненную орбиталь, т.е. происходит перераспределение электронной плотности.В органич.химии связь, образовавшуюся в результате координации 2-х нейтральных частиц, называют семиполярной. Фактически эта связь является ковалентной, соединяющей атомы, на которых при возникновении связи образуются положительный и отрицательный заряды.Под химич.связью обычно понимают совокупность сил, удерживающих два и более атома в устойчивой многоатомной системе. Образование химич.связи в отличие от межмолекулярного взаимодействия сопровождается существенной пересройкой электронных оболочек связывающихся атомов.
3.ГИБРИДИЗАЦИИ. ВАЛЕНТНЫЕ СОСОТОЯНИЯ АТОМА УГЛЕРОДА.Идея предсказания структурных формул молекулы формулируется в теории отталкивания следующим образом: пары электронов на валентной оболочке атома отталкивают друг друга и атомы располагаются таким образом, чтобы уменьшить отталкивание.В 1928-1931 гг Л.Полинг и Дж.Слейтер ввели понятие гибридизации атомных орбиталей. Гибридизация-метод математического комбинирования различных по форме и энергии орбиталей с образованием такого же количества новых, но уже одинаковых по форме и энергии гибридных орбиталей. Условием устойчивой гибридизации является энергетическая близость, достаточная близость и степень перекрывания.В соединениях атом углерода может находиться в одном из 3-х валентных состояний.Наиболее выгодна sp3-гибридизация, т.к. в полной мере реализуется принцип:-максимального перекрывания орбиталей;-максимального удаления всех орбиталей;Определение типа гибридизации (по структурной формуле):1. По количеству партнеров. Партнер-соседний атом и свободная электронная пара.-4 партнера соответствуют sp3-гибридизации (4б-связи);-3 партнера соответствуют sp2-гибридизации(связь двойная);-3 партнера соответствуют sp-гибридизации(связь тройная)2.Для атомов с 4-мя партнерами, среди которых электронная пара, учитывается гибридизация партнера. В случае sp2-гибридизации партнера для характеризуемого атома более выгодна sp2-гибридизация, т.к. реализуется возможность делокализации электронной пары по сопряженной системе и, а следствие, снижения энергии.С изменением типа гибридизации углеродного атома изменяются его свойства. При переходе от sp3-гибридизации к sp2-гибридизации изменяется форма гибридной орбитали.По характеру атомы углерода бывают:-первичные, соединенные с одним атомом углерода;-вторичные, соединенные с двумя атомами углерода;-третичные, соединенные с тремя атомами углерода;-четвертичные, соединенные с четырьмя атомами углерода Атом водорода называют:-первичным, если он соединен с первичным атомом углерода;-вторичным, если он соединен со вторичным атомом углерода;-третичным, если он соединен с третичным атомом углерода;-винильным, если он соединен с sp2-гибридизированным атомом углерода винильного фрагмента;-ацетиленовым, если он соединен с sp-гибридизированным атомом углерода (обуславливает кислотные свойства ацетиленов;-бензильным, если находится у атома углерода, связанного с ароматическим ядром.
4.КЛАССИФИК И НОМЕНКЛАТУРА ОРГАН СОЕД.Классификация- распределение органич. соединений по рядам, группам, классам в зависимости от их классификационных признаков. Классификация основана на структуре углеводородного скелета и характере функциональных групп.Классификационные признаки:1.структура углеродного скелета углеводородного фрагмента молекулы. Структура молекулы-определенная последовательность соединения атомов в молекуле и характер их связей. По этому признаку оргнанич.соединени я делятся на след.ряды:- ряд ациклических соединений;- ряд карбоциклических соединений;- ряд гетероциклических соединений;2.электронное строение молекулы.По этому признаку органич.соединения подразделяются на:- алифатические соединения;- ароматические соединения;3.характер функциональной группы, определяющей функциональный класс- группа соединений, объединяемых общей функциональной группой. При замене в углеводороде (R-II) атомов водорода на другие атомы или функциональные группы (Х) образуются новые классы органич.соединений (R-Х), характер которых определяют функциональные группы.Функциональная группа- гетероатом или группа атомов неуглеводородного характера, определяющих принадлежность к определенному функциональному классу и обуславливающая важнейшие свойства этого класса.Номенклатура органич.соединений.Номенклатура-это система правил построения названия соединения. Огромное число органич.соединений обуславливают сложность их номенклатуры. Предложено несколько различных систем номенклатуры:1.тривиальная номенклатура. Органич.соединения получили случайные названия, в которых отражались либо природные источники получения (яблочная кислота, муравьиная кислота, винный спирт), либо заметные свойства (гремучая кислота) и т.д.2.рациональная номенклатура связывает название вещества с его строением и классом. Для того, чтобы назвать соединение по рациональной номенклатуре, необходимо:-определить класс называемого соединения;-выбрать в соединении основу;-выбрать окружающие заместители;-составить название, начиная с названий простых заместителей к более сложным, заканчивая названием основы.Углеводородный радикал(заместитель)- остаток молекулы углеводорода, из которого формально удалили один или несколько атомов водорода, оставив свободными соответственно одну или несколько валентностей.Название углеводородных радикалов состоит из: префикса, корня, суффикса3.систематическая номенклатура. В 1957 г были приняты правила IUPAC.В рамках этой номенклатуры соединения рассматриваются как продукты усложнения нормальных предельных углеводородов либо замещенных циклов, получаемых путем замещения атомов водорода какими-либо структурными фрагментами. Характер заместителя указывается приставкой или окончанием. Для уточнения положения производится нумерация атомов основы.Способы построения названия: -выбрать основу, в качестве которой избирается самая длинная цепь углеродных атомов, в которой содержатся функциональные группы и кратные связи;-пронумеровать основу, начиная с наиболее замещенного конца. Начало нумерации определяет старшая функциональная группа, затем кратная связь и углеводородный заместитель.-составить название, включающее название заместителей в алфавитном порядке, название главной цепи, окончание, характерное для кратной связи и старшей функциональной группы. Положение заместителей и функциональной группы указывается цифрами, их количество – греческими числительными.
5.ОСН ПОНЯТИЯ О РЕАКЦИОННОЙ СПОСОБНОСТИ ОРГАН МОЛЕКУЛ.Химические реакции представляют собой процессы, в которых происходит перераспределение электронов. Направление и механизм реакции зависят от распределения электронов вступающих во взаимодействие молекул.В молекулах осуществляется взаимное влияние не связанных атомов, которое может реализовываться двумя путями. В связи с этим различают два механизма электронных смещений в молекуле: индуктивное влияние, мезомерный эффект или эффект сопряжения. Индукционный эффект состоит в передаче влияния путем последовательной поляризации б-связей и распространяется вдоль цепи связанных атомов по механизму электростатической индукции. Характерная особенность состоит в том, что все смещаемые электронные пары б-связей остаются на своих первоначальных октетах. Изменения касаются только их положения относительно связанных атомов. Направление индуктивного влияния определяют по частичному заряду, который приобретает заместитель (Х), его вызывающий.Сила индукционного эффекта зависит от природы заместителя. Электроотрицательность атома галогена возрастает от йода к фтору. Соответственно, усиливается вызываемый ими I эффект, и увеличиваются кислотные свойства галогенозамещенных карбоновых кислот.Еще одной характерной чертой индукционного эффекта является его быстрое затухание по цепи простых связей.Мезомерный эффект представляет собой еще дин механизм передачи взаимного влияния атомов, не связанных друг с другом. Под действием мезомерного эффекта происходит перераспределение электронной плотности в молекуле. В отличие от индуктивного влияния это перераспределение осуществляется не через систему б-связей, а главным образом с участием п- и p- электронов. Если при индуктивном влиянии б-электроны частично смещаются относительно исходных положений, то в случае мезомерного эффекта происходит частичное перемещение электронных пар в соседние октеты, вызывающее аналогичное отталкивание взаимодействующих электронных пар вдоль цепи п-связей вплоть до крайнего атома, на котором локализуется избыток электронной плотности.В отличие от индукционного мезомерный эффект передается по системе п-связей практически не ослабевая, стабилизируя молекулу и понижая ее систему. Особое положение в ряду различных видов мезомерного эффекта занимает б, п-сопряжение или сверхсопряжение. Перекрывание является частичным и неэффективным и происходит в силу поляризуемости б-молекулярной орбитали.
6.
Гомол. ряд метана. Изомерия.
Соед, сходн по хим св-ам, состав кот
отлич друг от друга на гр СН2,
назыв гомологами. Гомологи, располож
в порядке возраст их относит молекул
массы, образ гомологич ряд. Группы
СН2 назыв
гомологич разностью. Примером гомолог
ряда мож служить ряд предельн угл-ов
(алканов). Простейш его представит -
метан СН4.
Гомологами метана явл: этан С2Н6,
пропан С3Н8
и т. д. Формула любого послед гомолога
мож быть получена прибавлен к формуле
предыдущ угл-а гомологич разности.Состав
молекул всех членов гомологич ряда мож
быть выражен одной общ формулой. Для
предел угл-ов такой формулой будет
СnН2n+2 ,
где n -
число атомов углерода.
Изомерия
– явлен существов соед, кот имеют
одинаков состав (одинак молек формулу),
но разн строен. Такие соед назыв изомерами.
Различ
в порядке соед атомов в молекулах (в
хим строении) приводят к структурн
изомерии.
Строен структ изомеров отражается
структ формулами. В ряду алканов структ
изомерия проявл при содерж в цепи 4-х и
более атомов углерода, т.е. начиная с
бутанаС4Н10. Если
в молекулах одинак состава и одинак
хим строения возмож различ взаимн
располож атомов в пространстве, то
наблюд пространств
изомерия (стереоизомерия).
Алканы, начиная с этана H3C–СН3,
сущ в различ пространств формах
(конформациях),
обусловленных внутримолек вращ по
σ-связям С–С, и
проявляют поворотную
(конформац) изомерию.
В зависимости от строен цепи атомы
углерода, вход в ее состав, различ
следующ образом: атом С, связанный в
цепи только с 1 атомом С, назыв первичн,
с 2 - вторичн,
с 3 - третичн,
с 4 – четвертичн.
Рациональная Выбирается
один из атомов углеродной цепи, он
считается замещ метаном и относит него
строится назван «алкил1алкил2алкил3алкил4метан».
По номенклатуре ИЮПАК названия
алканов образ при помощи суффикса -ан путём
добавлен к соответств корню от назв
углеводорода. Выбирается наиболее
длинная неразветвлён углеводород цепь
так, чтобы у наибольш числа заместит
был минималь номер в цепи. В названии
соед цифрой указывают номер углеродного
атома, при котором находится замещающая
гр или гетероатом,
затем назван гр или гетероатома и назван
главной цепи. Если группы повтор, то
перечисляют цифры, указывающие их
положение, а число одинаковых групп
указывают приставками ди-, три-, тетра-.
Если группы неодинак, то их назв
перечисляются в алфавит порядке.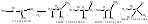
7.Природ источ алканов, значение. Синтез. В земной атмосфере метан присутств в очень небольш количествах (около 0,0001 %), он производится некоторыми археями (архебактериями), в частности, находящ в кишечн тракте крупн рогат скота. Промышл знач имеют месторождения низш алканов в форме природ газа, нефти и, вероятно, в будущем — газовых гидратов (найдены в областях вечной мерзлоты и под океанами). Также метан содержится в биогазе. Высш алканы содерж в кутикуле растений, предохраняя их от высых, паразитн грибков и мелк растительноядн тварей. Синтез Фишера — Тропша nCO +(2n+1)H2 →CnH2n+2 +nH2O!!!!Восстан галогенпроизводн алканов:При каталитич гидрировании в присутствии палладия галогеналканы превращаются в алканы:R—CH2Cl+H2 →R—CH3 +HCl!!Восстановлен иодалканов происход при нагреван последн с иодоводородной кислотой:R—CH2I + HI → R—CH3 + I2!!!Для восстановлен галогеналканов пригодны также амальгама натрия, гидриды металлов, натрий в спирте, цинк в соляной кислоте или цинк в спирте,!Восстановление спиртов:Восстановление спиртов приводит к образованию углеводородов, содержащих то же количество атомов С. Так, например, проходит реакция восстановления бутанола(C4H9OH), проходящую в присутствии LiAlH4. При этом выделяется вода.H3C—CH2—CH2—CH2OH → H3C—CH2—CH2—CH3 + H2O.Восстановление карбонильных соединений Реакция Кижнера—Вольфа:
![]()
Реакцию проводят в избытке гидразинав высококипящем растворителе в присутствииKOH.
Реакция Клемменсена:
![]()
Гидрирование непредельных углеводородов Из алкенов CnH2n +H2 →CnH2n+2.Из алкиновCnH2n-2+2H2→ CnH2n+2 Катализ реакц явл соед никеля,платиныилипалладия. Синтез Кольбе При электролизесолей карбоновых кислот, анион кислоты — RCOO− перемещается к аноду, и там, отдавая электрон превращается в неустойчивый радикал RCOO•, который сразу декарбоксилируется. Радикал R• стабилизируется путем сдваивания с подобным радикалом, и образуется R—R[10]. Например:2CH3COO− − 2e → 2[CH3COO•] → 2CH3• → C2H6 -////- 2C3H7COOK → {электролиз} → C6H14 Газификация твердого топлива Проходит при повышенной температуре и давлении. Катализатор — Ni:C+2H2 → CH4 Реакция Вюрца 2R—Br + 2Na = R—R + 2NaBr Реакция идёт в ТГФ при температуре −80 °C[11]. При взаимодействии R и R` возможно образование смеси продуктов (R—R, R`—R`, R—R`)
8.Хим св-ва алканов. Р. Замещения, механизм S. Галогенирование. Алканы имеют низкую химич активность. Это объясняется тем, что единич связи C—H и C—C относит прочны и их сложн разруш. Поскольку углеродн связи неполярны, а связи С—Н малополярны, оба вида связей малополяризуемы и относятся к σ-виду, их разрыв наиболее вероятен по гомолитич механизму, то есть с образован радикалов. А. относ. к инертным в хим. отнош. в-вам. Коновалов назвал их «хим. мертвецами». Старое наз. алканов- парафины. Причина хим. устойч.: высокая прочность σ-св. C-C,C-H, неполярн. св. C-C и низкая полярн. C-H (малая разница ЭО по шкале Полинга 2,5 и 2,1). Низкая поляризуем. связей. В обычных условиях на алканы не действ. конц. к-ты и щелочи, сильные ок-ли, щел. метал. Неполярн. св. могут расщ. при атаке своб. радикалами (р-ции замещения, крекинг, окисления (горение)). Легче разрыв.связь C-H т.к. она более доступна для атаки реагента. Для алканов х-ны р-ции радикальн. замещ-я. Основные стадии р-ций замещ.по радикальному м-му: Зарождение цепи– образ. своб. радикалов, стадия роста цепи- образов. радикал отрыв. ат. Н с образ. нового радикала алкильного RH +Х•→ HХ + R•. Радикальный центр большин. алкильных радик. имеет плоск. конфигур. Стабильность радик-в в ряду метильн., первичн., вторичн., третичн. слева направо увелич. Получен. радикалы взаимод. с мол-ми реагента, что прив. к образ. нового радик. и пр-та замещ. RH. Т.о. на кажд. стадии генер. радикал, что характ. для цепн. р-ций. Обрыв цепи: рекомбинация 2 одинак. или разн. радикалов. В-ва своб. реагир. со своб. радик., тормоз. радик. Р-ции- ингибиторы. Галогенирование алканов подчиняется правилу Марковника — в первую очередь галогенируется наименее гидрированый атом углерода. Галогенирование алканов проходит поэтапно — за один этап галогенируется не более одного атома водорода.
1)Галогенирование.
CnН2n+2 + F2 = CnF2n+2 +НF Условия: Темпер. >400˚С. Ат Cl связ.с любым ат. H, с кот. он сталкив. Соотнош. монохлоридов: первичных, вторичных, третичных состав. 75%:15%:8%. Хлориров. при более низких темпер. протек. с больш. избирател. Бромирование алканов протек. с высок. избирательн. (региоселективностью реакции). Ат. Br менее реакционно способен (ввиду больш. стабильн.) прояв. значит. селективность в отрыве ат. Н. Это соотн. с энерг. связи. При бромиров. пропана вторичный и первичный пр-ты соотн. 50:1. Усл. р-ции: 125˚С, свет. Радикал. иодиров. алканов эндотермично и прот. с трудом из-за низк. актив. радик. I. . Также р-ция обратима, образовавш. иодоводород восст-ет алкилиодид. Прямое фториров.– трудно контр. экзотерм. проц. Смесь фторалканов.Сульфохлорирование и сульфоокисление. Прямое сульфиров. А. протек. с трудом и сопровожд. ок-ем. Легче сульфируют. при совм. дейст. диоксида серы и хлора, или кислорода. Оба процесс. радик. замещ. иницир. УФ-облуч. или пероксидами.R-H + SO2 + Сl2→ R- SO2-Сl (алкансульфонилхлорид)+ HCl.(свет) М-м сходен с м-мом р-ции галагениров., но +2 стадии роста цепи еще. Сульфоокисление СН4+SО2 +О2+ СН3SО3Н + Н2О
9. Хим св-ва алканов Нитрирование (Реакция Коновалова)Алканы реагируют с 10% раствором азотной кислоты или оксидом азота N2O4 в газовой фазе при t 140° и небольшом давлен с образован нитропроизводных. Реакция так же подчин правилу Марковникова. RH + HNO3 = RNO2 + H2O т. е. 1 из атомов водорода замен на остаток NO2 (ни-трогруппа) и выдел вода.Особенност строен изомеров сильно отраж на течении этой реакции, так как легче всего она ведет к замещ на нитрогр атома H в остатке СИ (имеющемся лишь в некоторых изомерах), менее легко замещается водород в группе СН2 и еще труднее — в остатке СН3.Парафины довольно легко нитруются в газовой фазе при 150—475° С двуокисью азота или парами азотн ки-ты; при этом происходит частич и окислен. Нитрованием метана получ почти исключит нитрометан.Все имеющиеся данные указывают на свободнорадикальный механизм. В результате реакции образуются смеси продуктов. Азотная кислота при обыкновен t почти не действ на парафиновые углеводороды. При нагревании же действует главным образом как окислитель. Однако, как нашел М. И. Коновалов (1889), при нагревании азотн кислота действует отчасти и «нитрующим» образом; особенно хорошо идет реакция нитрован со слабой азотной кислотой при нагревании и повыш давлении. Реакция нитрования выраж уравнением.Последующие за метаном гомологи дают смесь различных нитропарафинов вследствие попутно идущего расщепления. При нитровании этана получаются нитроэтан СН3—СН2—NO2 и нитрометан СН3—NO2. Из пропана образуется смесь нитропарафинов.Нитрование парафинов в газовой фазе теперь осуществляется в промышленном масштабе. Нитрование. Нитруются азотной к-той по радик. м-му (NO2-радикал-нитрония). Нитров. протек. С СН3-СН3+HNО3=СН3-СН2-NО2+Н2О+СН3-СН2-ОН разрывом св-й C-C и сопровож. окисл-ем. Реакция Коновалова. Усл.: 110-140гр,10-20% HNO3.Полаг., что р-ция Коновалова –гомолитическ. процесс.
10.Хим св-ва алканов Р.алканов с О2. Горение. Алканы имеют низкую химич активность. Это объясняется тем, что единич связи C—H и C—C относит прочны и их сложн разруш. Поскольку углеродн связи неполярны, а связи С—Н малополярны, оба вида связей малополяризуемы и относятся к σ-виду, их разрыв наиболее вероятен по гомолитич механизму, то есть с образован радикалов. А. относ. к инертным в хим. отнош. в-вам. Коновалов назвал их «хим. мертвецами». Старое наз. алканов- парафины. Причина хим. устойч.: высокая прочность σ-св. C-C,C-H, неполярн. св. C-C и низкая полярн. C-H (малая разница ЭО по шкале Полинга 2,5 и 2,1). Низкая поляризуем. связей. В обычных условиях на алканы не действ. конц. к-ты и щелочи, сильные ок-ли, щел. метал. Неполярн. св. могут расщ. при атаке своб. радикалами (р-ции замещения, крекинг, окисления (горение)). Легче разрыв.связь C-H т.к. она более доступна для атаки реагента. Для алканов х-ны р-ции радикальн. замещ-я. Реакции окисления:Горение Основным химическим свойством предельных углеводородов, определяющих их использование в качестве топлива, является реакция горения. Пример:CH4 + 2O2 → CO2 + 2H2O + Q. Значение Q достигает 46 000 — 50 000 кДж/кг. В случае нехватки кислорода вместо углекислого газа получается угарный газ или уголь (в зависимости от концентрации кислорода).В общем виде реакцию горения алканов можно записать следующим образом: СnН2n+2 +(1,5n+0,5)O2 → nCO2 + (n+1)H2O. Окисление также может осуществляться воздухом. Процесс проводится в жидкой или газообразной фазе. В промышленности так получают высшие жирные спирты и соответствующие кислоты.Механизм реакций получения кислот путём каталитического окисления и расщепления алканов показан ниже на примере получения из бутана уксусной кислоты:
Практическую
значимость приобретают методы синтеза
высших жирных кислот из олефинов в
присутствии Со2(СО)8:
гидрокарбоксилирование при 145-165 °С и
5-30 МПа: RCH=СН2 +
СО + Н2О
-> RCH2CH2COOH;
гидрокарбоалкоксилирование при 165-175
°С и 5-15 МПа с последующим
гидролизомобразующегося эфира: Преимущества
процессов: малостадийность, высокие
выходы кислот; недостатки: довольно
жесткие условия, образование большого
количества (до 50%) кислот изостроения. ![]() Реакции
окисления —
основаны на окислении углеводородов
(реже — галогенпроизводных
углеводородов), содержащих кратные или
активированные C−H связи. В качестве
окислительных агентов
для алканов и циклоалканов используются
сильные неорганические
окислители: озон, перманганат
калия, оксид
хрома (VI), хромовая
кислота, диоксид
селена, а также пероксид
водорода и
некоторые пероксикислоты. Из-за
возможности более глубокого окисления,
метод имеет значение, как правило,
только для получения третичных спиртов.
Реакции
окисления —
основаны на окислении углеводородов
(реже — галогенпроизводных
углеводородов), содержащих кратные или
активированные C−H связи. В качестве
окислительных агентов
для алканов и циклоалканов используются
сильные неорганические
окислители: озон, перманганат
калия, оксид
хрома (VI), хромовая
кислота, диоксид
селена, а также пероксид
водорода и
некоторые пероксикислоты. Из-за
возможности более глубокого окисления,
метод имеет значение, как правило,
только для получения третичных спиртов.
![]()
11Гомол. ряд
этиленовых угл-дов.Алкены
(олефины) –
это углеводороды, в молекулах которых
содержатся атомы углерода, соединенные
между собой двойной связью
(непредельные
углеводороды ряда этилена). Простейший
представитель — этиленС2Н4,
общая формула гомологического ряда
этиленовых углеводородов СnН2n (при
п ≥ 2).Систематические названия олефинов
производятся от корней названий алканов
с заменой суффикса – ан →
–ен:
CН2Н4-этен
СН3Н6-пропен С4Н8-бутен С5Н10-пентен
С6Н12-гексен С7Н14-гептан. ИзомерияДля
алкенов характерны два вида структурной
изомерии.
Кроме изомерии, связанной со строением
углеродного скелета (как у алканов),
появляется изомерия, зависящая
от положения двойной связи в цепи.
Это приводит к увеличению числа изомеров
в ряду алкенов. Первые два члена
гомологического ряда алкенов -этилен
и пропиле) - изомеров не имеют и их
строение можно выразить так:
H2C==CH2 H2C==CH—CH3
этилен
пропилен
(этен) (пропен). Для углеводорода
С4H8возможны
три изомера:
![]() Первые
два отличаются между собой положением
двойной связи углеродной цепи, а третий
—характером цепи (изостроение). Однако
в ряду этиленовых углеводородов помимо
структурноq изомерии возможен еще один
вид изомерии — цис-,транс-изомерия/ Такая
изомерия характерна для соединений с
двойной связью. Если простая s-связь
допускает свободное вращение отдельных
звеньев углеродной цепи вокруг своей
оси, то вокруг двойной связи такого
вращения не происходит. Это и является
причиной появления геометрических(цис-,
транс-)изомеров.Геометрическая
изомерия —
один из видов пространственной
изомерии.
Изомеры, у которых одинаковые
заместители(при разных углеродных
атомах) расположены по одну сторону от
двойной связи, называют цис-изомерами,а
по разную —транс-изомерами:
Первые
два отличаются между собой положением
двойной связи углеродной цепи, а третий
—характером цепи (изостроение). Однако
в ряду этиленовых углеводородов помимо
структурноq изомерии возможен еще один
вид изомерии — цис-,транс-изомерия/ Такая
изомерия характерна для соединений с
двойной связью. Если простая s-связь
допускает свободное вращение отдельных
звеньев углеродной цепи вокруг своей
оси, то вокруг двойной связи такого
вращения не происходит. Это и является
причиной появления геометрических(цис-,
транс-)изомеров.Геометрическая
изомерия —
один из видов пространственной
изомерии.
Изомеры, у которых одинаковые
заместители(при разных углеродных
атомах) расположены по одну сторону от
двойной связи, называют цис-изомерами,а
по разную —транс-изомерами:
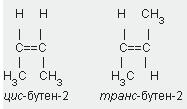 Цис-и транс-изомеры
отличаются не только пространственным
строением, но и многими физическими и
химическими свойствами. Транс-изомеры
более устойчивы, чем цис-изомеры.Номенклатура.
В
соответствии с правилами ИЮПАК при
построении алкенов
наиболее
длинная углеродная цепь, содержащая
двойную связь, получает название
соответствующего алкана, в котором
суффикс -ан заменён
на -ен.По
систематической номенклатуре названия
этиленовых углеводородов производят
заменой суффикса -ан в соответствующих
алканах на суффикс -ен (алкан — алкен,
этан — этен, пропан — пропен и т.д.).
Выбор главной цепи и порядок названия
тот же, что и для алканов. Однако в состав
цепи должна обязательно входить двойная
связь. Нумерацию цепи начинают с того
конца, к которому ближе расположена
эта связь. Например:
Цис-и транс-изомеры
отличаются не только пространственным
строением, но и многими физическими и
химическими свойствами. Транс-изомеры
более устойчивы, чем цис-изомеры.Номенклатура.
В
соответствии с правилами ИЮПАК при
построении алкенов
наиболее
длинная углеродная цепь, содержащая
двойную связь, получает название
соответствующего алкана, в котором
суффикс -ан заменён
на -ен.По
систематической номенклатуре названия
этиленовых углеводородов производят
заменой суффикса -ан в соответствующих
алканах на суффикс -ен (алкан — алкен,
этан — этен, пропан — пропен и т.д.).
Выбор главной цепи и порядок названия
тот же, что и для алканов. Однако в состав
цепи должна обязательно входить двойная
связь. Нумерацию цепи начинают с того
конца, к которому ближе расположена
эта связь. Например:
СH3 | H3C—CH2—C—CH==CH2 H3C—C==CH—CH—CH2—CH3 | | | CH3 CH3 CH3 3,3-диметилпентен-1 2,4-диметилгексен-2
Иногда используют и рациональные названия. В этом случае все алкеновые углеводороды рассматривают как замещенные-этилена:Н3С—СН==СН—CH2—СН3 метилэтилэтилен. Непредельные (алкеновые) радикалы называют тривиальными названиями или по систематической номенклатуре:Н2С==СН— - винил (этенил), Н2С==CН—СН2 - аллил (пропенил-2)
12Методы получения алкенов. Физические свойства алкенов похожи на свойства алканов, хотя все они имеют несколько более низкие температуры плавления и кипения, чем соответствующие алканы. Например, пентан имеет температуру кипения 36 °С, а пентен-1 — 30 °С. При обычных условиях алкены С2 - С4 — газы. С5 – С15 — жидкости, начиная сC16 — твердые вещества. Алкены не растворимы в воде, хорошо растворимы в органических растворителях.В природе алкены встречаются редко. Поскольку алкены являются ценным сырьем для промышленного органического синтеза, разработаны многие способы их получения.1. Основным промышленным источником алкенов служит крекинг алканов, входящих в состав нефти:
|
|
t |
|
|
С8Н18 |
→ |
С4Н10 + С4Н8 |
Крекинг протекает по свободнорадикальному механизму при высоких температурах (400-700 °С).2. Другой промышленный способ получения алкенов - дегидрирование алканов:
|
|
t, Cr2O3 |
|
|
СН3-СН2-СН3 |
→ |
СН3-СН=СН2 + Н2 |
3. В лабораторных условиях алкены получают по реакциям отщепления (элиминирования), при которых от соседних атомов углерода отщепляются два атома или две группы атомов, и образуется дополнительная p-связь. К таким реакциям относятся следующие.1) Дегидратация спиртов (отщепление воды)происходит при их нагревании с водоотнимающими средствами, например с серной кислотой при температуре выше 150 °С:
|
|
H2SO4 |
|
|
СН3-СН2-ОН |
→ |
СН2=СН2 + Н2О |
2) Отщепление галогеноводородов проводят при действии спиртовых растворов щелочей на моноалкилгалогениды:
|
|
С2Н6ОН |
|
|
СН3-СН2-СНВr-СН3 + КОН |
→ |
СН3-СН=СН-СН3 + КВr + Н2О |
При отщеплении Н2O от спиртов, НВr и HCl от алкилгалогенидов атом водорода преимущественно отщепляется от того из соседних атомов углерода, который связан с наименьшим числом атомов водорода (от наименее гидрогенизированного атома углерода). Эта закономерность носит название правила Зайцева. Гидрирование В присутствии металлических катализаторов (Pt, Ni) алкины присоединяют водород с образованием алкенов (разрывается первая π-связь), а затем алканов (разрывается вторая π-связь):
![]() При
использовании менее активного
катализатора [Pd/CaCO3/Pb(CH3COO)2] гидрирование
останавливается на стадии образования
алкенов. Участие
вторичных
спиртов при образовании алкена возможны
два направления реакции, преимущественное
направление то, при котором в процессе
конденсации
отщепляется
водород от наименее гидрогенизированного
атома углерода, т.е. окруженного меньшим
количеством атомов водорода.
При
использовании менее активного
катализатора [Pd/CaCO3/Pb(CH3COO)2] гидрирование
останавливается на стадии образования
алкенов. Участие
вторичных
спиртов при образовании алкена возможны
два направления реакции, преимущественное
направление то, при котором в процессе
конденсации
отщепляется
водород от наименее гидрогенизированного
атома углерода, т.е. окруженного меньшим
количеством атомов водорода.
13Хим
св-ва алкенов. Присоединение. Характер
углерод – углеродной связи определяет
тип химических реакций, в которые
вступают органические вещества. Наличие
в молекулах этиленовых углеводородов
двойной углерод – углеродной связи
обуславливает следующие особенности
этих соединений:
– наличие двойной
связи позволяет отнести алкены к
ненасыщенным соединениям. Превращение
их в насыщенные возможно только в
результате реакций присоединения, что
является основной чертой химического
поведения олефинов;– двойная связь
представляет собой значительную
концентрацию электронной плотности,
поэтому реакции присоединения носят
электрофильный характер;– двойная
связь состоит из одной ![]() -
и одной
-
и одной ![]() -связи,
которая достаточно легко поляризуется
Реакции
присоединения:
-связи,
которая достаточно легко поляризуется
Реакции
присоединения:
Гидрирование (присоединение водорода)Алкены взаимодействуют с водородом при нагревании и повышенном давлении в присутствии катализаторов (Pt, Pd, Ni и др.) с образованием алканов:
![]()
Гидрирование алкенов – реакция, обратная дегидрированию алканов. Согласнопринципу Ле Шателье, гидрированию благоприятствует повышенное давление, т.к. эта реакция сопровождается уменьшением объёма системы. Присоединение водорода к атомам углерода в алкенах приводит к понижению степени их окисления, поэтому гидрирование алкенов относят к реакциям восстановления. Эта реакция используется в промышленности для получения высокооктанового топлива. Реакции электрофильного присоединения — р. присоединения, в которых атаку на начальной стадии осуществляет электрофил — частица, заряженная положительно или имеющая дефицит электронов. На конечной стадии образующийся карбкатион подвергается нуклеофильной атаке. В органической химии чаще всего атакующей электрофильной частицей является протон H+.Несмотря на общность механизма различают реакции присоединения по связи углерод—углерод и углерод—гетероатом.Общий вид реакций присоединения по двойной связи углерод-углерод: Реакции электрофильного присоединения распространены среди алкенов и алкинов и широко используются в промышленном химическом производстве и лабораторных синтезах.
![]()
![]() Реакции
с
Н-электрофилами
(сильными
кислотами). Алкены реагируют с сильными
кислотами. Во многих случаях происходит
присоединение кислоты по двойной связи:
RCH
= CHR+H-a)U->
RCH2-CHR
где Х=С1, Br, I, OS03H и др.Хорошо изучены
реакции присоединения галогенводородов
(гидрогалогенирование) и серной кислоты.
Присоединение галогенводородов
происходит как в газовой фазе, так и в
растворах, на скорость реакции значительно
влияет присутствие солей тяжелых
металлов.В случае несимметричных
алкенов возможны два направления
присоединения кислоты. Эти реакции
подробно изучил В. В.Мар-ковников и
пришел к выводу, что в большинстве
случаев направление присоединения
предопределяется строением алкена.
Протон присоединяется к тому углеродному
атому, у которого меньше углеводородных
заместителей (к «более гидрированному»)
— правило
Марковникова
(1870): RCH = CH2 + HX —? RCH-CHs. Эта направленность
легко объясняется классической
электронной теорией. Молекула
несимметрично замещенного алкена
является поляризованной, а алкильные
группы как электронодонорные запо
местители определяют наиболее вероятное
место присоединения протона. Так как
алкильные группы являются слабыми
донорами, направленность реакции
присоединения может меняться в
зависимости от условий реакций,
растворителя, соотношения концентраций
компонентов, температуры.
Гидратация алкенов
– это присоединение воды по двойной
С=С-связи:
Реакции
с
Н-электрофилами
(сильными
кислотами). Алкены реагируют с сильными
кислотами. Во многих случаях происходит
присоединение кислоты по двойной связи:
RCH
= CHR+H-a)U->
RCH2-CHR
где Х=С1, Br, I, OS03H и др.Хорошо изучены
реакции присоединения галогенводородов
(гидрогалогенирование) и серной кислоты.
Присоединение галогенводородов
происходит как в газовой фазе, так и в
растворах, на скорость реакции значительно
влияет присутствие солей тяжелых
металлов.В случае несимметричных
алкенов возможны два направления
присоединения кислоты. Эти реакции
подробно изучил В. В.Мар-ковников и
пришел к выводу, что в большинстве
случаев направление присоединения
предопределяется строением алкена.
Протон присоединяется к тому углеродному
атому, у которого меньше углеводородных
заместителей (к «более гидрированному»)
— правило
Марковникова
(1870): RCH = CH2 + HX —? RCH-CHs. Эта направленность
легко объясняется классической
электронной теорией. Молекула
несимметрично замещенного алкена
является поляризованной, а алкильные
группы как электронодонорные запо
местители определяют наиболее вероятное
место присоединения протона. Так как
алкильные группы являются слабыми
донорами, направленность реакции
присоединения может меняться в
зависимости от условий реакций,
растворителя, соотношения концентраций
компонентов, температуры.
Гидратация алкенов
– это присоединение воды по двойной
С=С-связи:
![]() Непосредственно
вода с алкенами не реагирует. Нужны
электрофильные частицы, например Н+,
а их в воде мало. Поэтому используют
катализатор
(источник
протонов), обычно серную
кислоту.
Присоединение протекает по правилу
Марковникова.
Пример:
Непосредственно
вода с алкенами не реагирует. Нужны
электрофильные частицы, например Н+,
а их в воде мало. Поэтому используют
катализатор
(источник
протонов), обычно серную
кислоту.
Присоединение протекает по правилу
Марковникова.
Пример:
![]() Промышленные
синтезы:
Промышленные
синтезы:
![]() Выход
в этой реакции 3–5%, поэтому смесь
пропускают через катализатор
Выход
в этой реакции 3–5%, поэтому смесь
пропускают через катализатор ![]() 100
раз.
100
раз.
![]() В
лабораторной практике этот метод обычно
применяют для синтеза третичных спиртов
(группа ОН связана с третичным атомом
углерода). При образовании вторичных
спиртов (группа ОН связана со вторичным
атомом углерода) велико содержание
побочных продуктов.
В
лабораторной практике этот метод обычно
применяют для синтеза третичных спиртов
(группа ОН связана с третичным атомом
углерода). При образовании вторичных
спиртов (группа ОН связана со вторичным
атомом углерода) велико содержание
побочных продуктов.
14Хим
св-ва алкенов. С-электрофилы. Характер
углерод – углеродной связи определяет
тип химических реакций, в которые
вступают органические вещества. Наличие
в молекулах этиленовых углеводородов
двойной углерод – углеродной связи
обуславливает следующие особенности
этих соединений:
– наличие двойной
связи позволяет отнести алкены к
ненасыщенным соединениям. Превращение
их в насыщенные возможно только в
результате реакций присоединения, что
является основной чертой химического
поведения олефинов;– двойная связь
представляет собой значительную
концентрацию электронной плотности,
поэтому реакции присоединения носят
электрофильный характер;– двойная
связь состоит из одной ![]() -
и одной
-
и одной ![]() -связи,
которая достаточно легко поляризуется.
Реакции
с электрофилами Изонитрилы
реагируют с электрофильными реагентами
с образованием продуктов 1,1-присоединения
к атому углерода, при этом Изонитрилы
бурно реагируют с галогенами - хлором,
бромом и даже йодом, образуя
иминокарбонилдигалогениды, в синтетической
практике реакцию проводят при охлаждении:
R—N≡C
+ Hal2
-связи,
которая достаточно легко поляризуется.
Реакции
с электрофилами Изонитрилы
реагируют с электрофильными реагентами
с образованием продуктов 1,1-присоединения
к атому углерода, при этом Изонитрилы
бурно реагируют с галогенами - хлором,
бромом и даже йодом, образуя
иминокарбонилдигалогениды, в синтетической
практике реакцию проводят при охлаждении:
R—N≡C
+ Hal2 ![]() RNC(Hal)2.
Способность
алкенов вступать в реакцию с электрофильными
реагентами обусловлена повышенной
электронной плотностью в области
двойной связи.
Исследователи
разработали
родиевые катализаторы, которые впервые
позволили обеспечить как высокое число
каталитических циклов на моль
катализатора, так и высокую
селективность в
реакции получения хиральных альдегидов
из ахиральных алкенов.
Исследователи смогли превратить реакцию
гидроформилирования, крупнотоннажное
производство, объёмом в 3,5 млн. кг
производимого продукта в год, в
энантиоселективный процесс, используя
хиральные бис-3,4-диазафосфолановые
лиганды. Дополняя результаты группа
профессоров– конденсировали рацемические
бис-3,4-диазафосфоланы с энантиомерно-чистыми
аминами. Полученные диастереомерные
бензамиды были разделены с помощью
флэш-хроматографии. Другие специалисты
тестировали новые лиганды в
родий-катализируемой реакции
гидроформилирования стирола, аллилцианида
и винилацетата. Реакция проводилась в
мягких условиях (20-500 psi СО/Н2,
смесь, называемая "синтез-газом",
40-120 °C). При этом для всех трёх субстратов
катализатор показал высокую активность
и селективность. При температуре 60 °C
и давлении 500 psi, лучший лиганд (см. рис.)
показал энантиоселективность от 87 до
95% и число циклов порядка 3000 в час.
Гипогалогенирование. Присоединение
к олефинам гипогалогенитных кислот и
их эфиров осуществляется согласно
следующей реакции: CH2 = СН2 + НОС1
RNC(Hal)2.
Способность
алкенов вступать в реакцию с электрофильными
реагентами обусловлена повышенной
электронной плотностью в области
двойной связи.
Исследователи
разработали
родиевые катализаторы, которые впервые
позволили обеспечить как высокое число
каталитических циклов на моль
катализатора, так и высокую
селективность в
реакции получения хиральных альдегидов
из ахиральных алкенов.
Исследователи смогли превратить реакцию
гидроформилирования, крупнотоннажное
производство, объёмом в 3,5 млн. кг
производимого продукта в год, в
энантиоселективный процесс, используя
хиральные бис-3,4-диазафосфолановые
лиганды. Дополняя результаты группа
профессоров– конденсировали рацемические
бис-3,4-диазафосфоланы с энантиомерно-чистыми
аминами. Полученные диастереомерные
бензамиды были разделены с помощью
флэш-хроматографии. Другие специалисты
тестировали новые лиганды в
родий-катализируемой реакции
гидроформилирования стирола, аллилцианида
и винилацетата. Реакция проводилась в
мягких условиях (20-500 psi СО/Н2,
смесь, называемая "синтез-газом",
40-120 °C). При этом для всех трёх субстратов
катализатор показал высокую активность
и селективность. При температуре 60 °C
и давлении 500 psi, лучший лиганд (см. рис.)
показал энантиоселективность от 87 до
95% и число циклов порядка 3000 в час.
Гипогалогенирование. Присоединение
к олефинам гипогалогенитных кислот и
их эфиров осуществляется согласно
следующей реакции: CH2 = СН2 + НОС1 ![]() СН2Сl-СН2ОН.
Механизм процесса
оксосинтеза
включает стадии активации и превращения
водорода и алкена. Активация водорода
приводит к образованию гидрокарбонила
кобальта. При взаимодействии алкена
с ненасыщенным гидрокарбонилом кобальта
образуется пи-комплекс, который может
превратиться в два различных
сигма-кобальторганических соединений
в зависимости от направления нуклеофильного
присоединения гидридного лиганда и
кобальта с карбонильными лигандами.
Далее кобальталкильные соединения
подвергаются внедрению карбонильной
группы из внутренней сферы комплекса
с заполнением освободившегося места
молекулой оксида углерода. Завершает
каталитический цикл стадия гидрогенолиза
с образованием альдегида линейного
или разветвленного строения.
СН2Сl-СН2ОН.
Механизм процесса
оксосинтеза
включает стадии активации и превращения
водорода и алкена. Активация водорода
приводит к образованию гидрокарбонила
кобальта. При взаимодействии алкена
с ненасыщенным гидрокарбонилом кобальта
образуется пи-комплекс, который может
превратиться в два различных
сигма-кобальторганических соединений
в зависимости от направления нуклеофильного
присоединения гидридного лиганда и
кобальта с карбонильными лигандами.
Далее кобальталкильные соединения
подвергаются внедрению карбонильной
группы из внутренней сферы комплекса
с заполнением освободившегося места
молекулой оксида углерода. Завершает
каталитический цикл стадия гидрогенолиза
с образованием альдегида линейного
или разветвленного строения.
15Хим
св-ва алкенов. Р. Окисления.Характер
углерод – углеродной связи определяет
тип химических реакций, в которые
вступают органические вещества. Наличие
в молекулах этиленовых углеводородов
двойной углерод – углеродной связи
обуславливает следующие особенности
этих соединений:
– наличие двойной
связи позволяет отнести алкены к
ненасыщенным соединениям. Превращение
их в насыщенные возможно только в
результате реакций присоединения, что
является основной чертой химического
поведения олефинов;– двойная связь
представляет собой значительную
концентрацию электронной плотности,
поэтому реакции присоединения носят
электрофильный характер;– двойная
связь состоит из одной ![]() -
и одной
-
и одной ![]() -связи,
которая достаточно легко поляризуется.
Реакция
Вагнера (окисление по Вагнеру,
перманганатная
проба) Окисление
органических соединений, содержащих
двойную связь, действием 1–3%-го раствора
перманганата калия (1887)
в цис-a-гликоли
в щелочной среде (считается положительной,
если раствор перманганата быстро
обесцвечивается в кислой среде или
буреет в щелочной и нейтральной):
-связи,
которая достаточно легко поляризуется.
Реакция
Вагнера (окисление по Вагнеру,
перманганатная
проба) Окисление
органических соединений, содержащих
двойную связь, действием 1–3%-го раствора
перманганата калия (1887)
в цис-a-гликоли
в щелочной среде (считается положительной,
если раствор перманганата быстро
обесцвечивается в кислой среде или
буреет в щелочной и нейтральной):
![]() Эпоксиды —
насыщенные трехчленные гетероциклы,
содержащие в цикле один кислородный
атом[1].
Эпоксиды являются циклическими простыми
эфирами, однако вследствие напряженности
трехчленного цикла обладают высокой
реакционной способностью в реакциях
раскрытия цикла. Низшие эпоксиды —
за исключением газообразной при
нормальных условиях окиси
этилена —
жидкости с эфирным запахом, хорошо
растворимые в органических растворителях,
температуры кипения эпоксидов несколько
выше температур кипения простых эфиров
с близкими молекулярными массами.
Наиболее
общими методами синтеза эпоксидов
являются селективное окисление алкенов
(эпоксидирование) и циклизация при
дегидрогалогенировании галогенгидриновпод
действием оснований.Лабораторным
методом эпоксидирования алкенов
является реакция
Прилежаева—
взаимодействие алкенов с перкарбоновыми
кислотами в
инертных неполярных или слабополярных
растворителях:
Эпоксиды —
насыщенные трехчленные гетероциклы,
содержащие в цикле один кислородный
атом[1].
Эпоксиды являются циклическими простыми
эфирами, однако вследствие напряженности
трехчленного цикла обладают высокой
реакционной способностью в реакциях
раскрытия цикла. Низшие эпоксиды —
за исключением газообразной при
нормальных условиях окиси
этилена —
жидкости с эфирным запахом, хорошо
растворимые в органических растворителях,
температуры кипения эпоксидов несколько
выше температур кипения простых эфиров
с близкими молекулярными массами.
Наиболее
общими методами синтеза эпоксидов
являются селективное окисление алкенов
(эпоксидирование) и циклизация при
дегидрогалогенировании галогенгидриновпод
действием оснований.Лабораторным
методом эпоксидирования алкенов
является реакция
Прилежаева—
взаимодействие алкенов с перкарбоновыми
кислотами в
инертных неполярных или слабополярных
растворителях:
![]()
Эпоксидирование
алкенов может осуществляться и под
действием других пероксидных соединений
(трет-бутилгидропероксид, перекись
водородав щелочной среде при
эпоксидировании α,β-непредельных
карбонильных соединений), в промышленности
этиленоксид получают каталитическим
окислением этилена кислородом воздуха.Окисление
с разрывом С–С-связей.
Реактивы, используемые для окисления:
KMnO4—H2SO4,
K2Cr2O7—H2SO4,
CrO3—CH3COOH.
Из неорганической химии известно, что
все они – весьма сильные окислители.
Продукты окисления – кетоны R2C=O,
кислоты RCOOH и углекислый газ. Рассмотрим:1)
RCH=CH2 ![]() [RCHO
+ CH2O]
[RCHO
+ CH2O] ![]() RCOOH
+ CO2;
В квадратных скобках указаны альдегиды
RCHO, неустойчивые в условиях реакции и
окисляющиеся до кислот (все, кроме
муравьиного альдегида СН2О):RCHO
RCOOH
+ CO2;
В квадратных скобках указаны альдегиды
RCHO, неустойчивые в условиях реакции и
окисляющиеся до кислот (все, кроме
муравьиного альдегида СН2О):RCHO ![]() RCOOH.
Муравьиный альдегид окисляется до
СО2:Н2СО
RCOOH.
Муравьиный альдегид окисляется до
СО2:Н2СО ![]() СО2 +
Н2О.
При окислении трехокисью
хрома в уксусном ангидриде
происходит окисление метильной группы
алкиларенов до альдегидной; дальнейшему
окислению до кислоты препятствует
образование диацетата, который устойчив
в этих условиях. Катализируемый кислотой
гидролиз в водном спирте приводит к
ароматическому альдегиду:
СО2 +
Н2О.
При окислении трехокисью
хрома в уксусном ангидриде
происходит окисление метильной группы
алкиларенов до альдегидной; дальнейшему
окислению до кислоты препятствует
образование диацетата, который устойчив
в этих условиях. Катализируемый кислотой
гидролиз в водном спирте приводит к
ароматическому альдегиду:
![]()
16.ХИМ СВ-ВА
АЛКЕНОВ. Р.ЦИКЛОПРИСОЕД.
Энергия двойной С-С связи в этилене
(146 ккал/моль) оказывается значительно
более низкой, чем удвоенная энергия
одинарной С-С-связи в этане (2 88=176
ккал/моль).
![]() -Связь
С-С в этилене прочнее
-Связь
С-С в этилене прочнее![]() -связи,
поэтому реакции алкенов, сопровождающиеся
разрывом -связи с образованием двух
новых простых
-связи,
поэтому реакции алкенов, сопровождающиеся
разрывом -связи с образованием двух
новых простых![]() -связей,
представляют собой термодинамически
благоприятный процесс. Так, например,
в газовой фазе согласно расчетным
данным все приведенные ниже реакции
являются экзотермическими со значительной
отрицательной энтальпией, независимо
от их реального механизма.
-связей,
представляют собой термодинамически
благоприятный процесс. Так, например,
в газовой фазе согласно расчетным
данным все приведенные ниже реакции
являются экзотермическими со значительной
отрицательной энтальпией, независимо
от их реального механизма.
![]()
![]()
![]()
![]()
Циклоприсоединение, р-ции, протекающие с образованием нового цикла из двух реагирующих молекул сопровождаются общим уменьшением кратности связей.
CH2=CH-CH3 + Br2 --(свет)--> CH2=CH-CH2Br + HBr (аллильное галогенирование)
17.АЦЕТИЛЕН,
ИЗОМЕРИЯ, НОМЕНКЛАТУРА.Алкины –
это углеводороды связаны друг с другом
тройной связью. Общая формула:
CnH2n–2.Особенности
алкинов:1)длина связи в алкинах равна
0,120 нм;2)каждый атом углерода в состоянии
sp-гибридизации связан с двумя другими
атомами;3)может присоединять еще два
атома. Простейшим
алкином является этин
(ацетилен)Гомологический
ряд:Этин:
C2H2,
Пропин:
C3H4,
Бутин:
C4H6,
Пентин:
C5H8,
Гексин:
C6H10,
Гептин:
C7H12,
Октин:
C8H14,
Нонин:
C9H16,
Децин:
C10H18
Сущ-ет 2 типа изомерии:1)изомерия
положения тройной связи;2)изомерия
цепи. Этин
и пропин –изомеров
не имеют.Для бутинов возможен
только один вид изомерии –
![]() изомерия
положения тройной связи.
Существует
2 типа номенклатуры: 1) международная
номенклатура: этин; пропин; 2) рациональная
номенклатура: ацетилен; метиацетилен.Физ.
свойства алкинов: 1) С2Н2…С4Н6 –
газы; 2) С5Н8…С15Н28 –
жидкости; 3) С16Н30…
– твердые вещества; 4) плохо растворимы
в воде.Хим. свойства алкинов: обладают
большой реакционной способностью,
харак-тся реакцией присоединения,
тройная связь содержит две
π-связи.Атомы углерода в
ацетилене sp-гибридизованы. Они связаны
одной
изомерия
положения тройной связи.
Существует
2 типа номенклатуры: 1) международная
номенклатура: этин; пропин; 2) рациональная
номенклатура: ацетилен; метиацетилен.Физ.
свойства алкинов: 1) С2Н2…С4Н6 –
газы; 2) С5Н8…С15Н28 –
жидкости; 3) С16Н30…
– твердые вещества; 4) плохо растворимы
в воде.Хим. свойства алкинов: обладают
большой реакционной способностью,
харак-тся реакцией присоединения,
тройная связь содержит две
π-связи.Атомы углерода в
ацетилене sp-гибридизованы. Они связаны
одной![]() и
двумя
и
двумя![]() связями,
макс. плотности к-рых расположены в
двух взаимно перпендикулярных областях,
образуя цилиндрич. облако
связями,
макс. плотности к-рых расположены в
двух взаимно перпендикулярных областях,
образуя цилиндрич. облако![]() электронной
плотности;
за его пределами находятся атомы Н.
Электронное строение ацетилена H:С:::С:H
Углеродные
атомы ацетилена, находясь в состоянии
sp-гибридизации, отличаются, как известно,
повышенной электроотрицательностью
. Поэтому электронная плотность связи
С—H несколько смещена в сторону углерода
и атом водорода приобретает некоторую
подвижность.
электронной
плотности;
за его пределами находятся атомы Н.
Электронное строение ацетилена H:С:::С:H
Углеродные
атомы ацетилена, находясь в состоянии
sp-гибридизации, отличаются, как известно,
повышенной электроотрицательностью
. Поэтому электронная плотность связи
С—H несколько смещена в сторону углерода
и атом водорода приобретает некоторую
подвижность.
18. Способы получения алкинов.Синтез.1. Карбидный метод (Ф.Велер, 1862 г.):СаС2 + 2Н20 НС ≡ СН + Са(ОН)2 Карбид кальция можно рассматривать как металлорганическое соединение, содержащее сильно полярную связь углерод - металл:
Аналогично реакция протекает для карбидов стронция, бария. Карбид магния в этих условиях образует пропин:
Mg2C3 + 4Н20 СН3С ≡ СН + Mg (OH)2 2. Из простых веществ (М.Бертло, 1860 г.):
2С
+ Н2
НС
≡ СН 3.
Пиролиз
(1000-
1200°С):
2СН4
 НС ≡ СН + ЗН2
-//- СН2
=
СН2
НС ≡ СН + ЗН2
-//- СН2
=
СН2
 НС ≡ СН + Н24.
Реакции
отщепления. 4.1
Дегалогенирование:
НС ≡ СН + Н24.
Реакции
отщепления. 4.1
Дегалогенирование:
5. Алкилирование алкинов (ж.И. Иоцич, 1902 г).
6. Получение из природных источников.Соединения с сопряженными тройными связями выделены из сложноцветных, зонтичных растений. Например, из подсолнечника выделен углеводород желтого цвета -тридецен-1-пентаин-3,5,7,9,11:
19.Хим.св.алкинов. Р присоед: гидрирование; взаим.с электрофильными реагентами; о Марковников. Алки́ны — углеводороды, содержащие тройную связь между атомами углерода, образующие гомологический ряд с общей формулой CnH2n-2. Атомы углерода при тройной связи находятся в состоянии sp-гибридизации.Для алкинов характерны реакции присоединения. В отличие от алкенов, которым свойственны реакции электрофильного присоединения, алкины могут вступать также и в реакции нуклеофильного присоединения. Это обусловлено значительным s-характером связи и, как следствие, повышенной электроотрицательностью атома углерода. Кроме того, большая подвижность атома водорода при тройной связи обуславливает кислотные свойства алкинов в реакциях замещения.Обусловлены С-Сπ связями (присоединение) и Сsp- Н связью (кислотные свойства).Электрофильное присоединение. Алкины в реакциях с электрофильными реагентами взаимодействуют аналогично алкенам, по тому же механизму электрофильного присоединенияк С-Сπ связи. Однако реакции этого типа идут труднее из-за компактной π-МО и более прочной С-Сπ связи в алкинах по сравнению с алкенами. т.о. Число электрофильных реагентов, способных присоединяться к алкинам, значительно уменьшается за счёт исключения слабых электрофилов.Галогены (Сl2,Вr2,I2) присоед.к алкинам, но с меньшей ск.,чем к алкенам; обычно присоед.две молекулы калогена:
CH3 ⁄ ⁄ Br
СН3-С≡ →Вr2 C═C
∕ ⁄ H
Вr Присоед.галогеноводородов. Они присоед.к алкинам так же как и к алкенам,но алкины могут присоед.одну или две молекулы галогеноводорода: обе молекулы галогеноводорода присоед.по правилу Марковникова
/Br
СН3—С≡СН +Вr→СН3—С=СН2→HВr СН3--С--СН3
⁄ /
Вr Вr
2-бромпропен 2,2-дибромпропан
20.Хим св-ва Алки́ны — углеводороды, содержащие тройную связь между атомами углерода, образующие гомологический ряд с общей формулой CnH2n-2. Атомы углерода при тройной связи находятся в состоянии sp-гибридизации.Для алкинов характерны реакции присоединения. В отличие от алкенов, которым свойственны реакции электрофильного присоединения, алкины могут вступать также и в реакции нуклеофильного присоединения. Это обусловлено значительным s-характером связи и, как следствие, повышенной электроотрицательностью атома углерода. Кроме того, большая подвижность атома водорода при тройной связи обуславливает кислотные свойства алкинов в реакциях замещения.Реакции присоединения:1) присоединение водорода (гидрирование). На I ступени образуются алкены, на II ступени – алканы.2) присоединение галогенов (галогенирование). HC≡CH + HCl → CH2=CHCl → CH3-CHCl2; На I ступени образуются дигалогеналкены, на II – тетрагалогеналканы.Реакция алкинов с бромной водой – качественная реакция на алкины. Бромная вода обесцвечивается;3) присоединение галогеноводородов (гидрогалогенирование).На I ступени образуются моногалогеналкены, на II – дигалогеналканы;4) присоединение воды (гидратация).Ацетилен образует альдегид, его гомологи – кетоны (реакция М.Г. Кучерова): Явление, когда вещество может существовать в виде нескольких изомерных форм, легко переходящих друг в друга и находящихся в динамическом равновесии, называют таутомерией. Переходящие друг в друга формы называются таутомерами, а их взаимный переход - таутомерным превращением. Таутомерные превращения характерны для многих классов органических соединений. Изложение материала, затрагивающего таутомерные системы, безусловно должно опираться на такие понятия, как взаимное влияние атомов в молекулах, скорость химических реакций, динамическое равновесие, влияние растворителей на состояние равновесия, термодинамическая устойчивость органических веществ, механизмы органических реакций. Поэтому изучение данного материала позволит представить органическую химию как целостную науку.Понятие таутомерии сформировалось еще в XIX веке, причем формирование его происходило достаточно обычным для химии методом - методом проб и ошибок. В 1865 году Ф. Кекуле предложил для бензола циклическую формулу с чередующимися двойными и одинарными связями. Эта формула предусматривала возможность существования следующих изомеров:ЭЛЬТЕКОВА ПРАВИЛО: ненасыщенные спирты с группой ОН у атома углерода, при к-ром имеется двойная связь (см. Енолы), нестойки и превращаются в карбонильные соед. (альдегиды или кетоны): Эльтекова правило в определенной мере справедливо лишь для простейших енолов. Известны многочисленные достаточно стабильные енолы[напр., соед. ф-лы I, СН3С(ОН) = =CHC(О)Ph, HOCH = C(COOC2H5)2 содержат соотв. 100, 99 и 94% енольной формы], а также соед., в к-рых енольная и карбонильная формы находятся в динамич. равновесии (см. Таутомерия).
21.Хим. св-ва алкинов.Кислот. св-ва. Ацетилениды Mg, Na, Li часто используют в орг-ком синтезе для введения этинильной группы. При взаимод. с алкилиру-ющими агентами обр. алкилацетилены (метилацетилен, 1-бутин, 1-пентин): RX + МССН → RCCH + MX.Ацетиленид Cu (I) — красно-кор., черные кристаллы. В сухом виде детонируют при ударе или нагреве, в отсутствие О2 не образуется газообр. в-в. Образуется в виде осадка при пропускании ацетилена в аммиачные р-ры солей меди(I). C2H2 + 2CuCl → Cu2C2 + 2HCl. Ацетиленид Ag — оч. неустойч. белое кристал. взрывчатое в-во. Пропустим через аммиач. р-р оксида Ag ацетилен. Выпадает осадок беловатого цвета - ацетиленид серебра.
ИОЦИЧА РЕАКЦИЯ
Реакция Фаворского1. Ацетилен-алленовая перегруппировка.1888 г. получил бутин-1 дегидрогалогенированием 2,2-дихлорбутана под действием спиртового р-ра КОН в ампуле при 170 °C. Вместо бутина-1 был получен бутин-2.CH3-CH2-C≡CH ↔ [CH3-CH=C=CH2] ↔ CH3-C≡C-CH3 2. Присоединение карбонильных соединений к алкинам.В присутствии сил. оснований алкины с концевой 3й связью способны присоед. карбонил. соединения с обр. спиртов:
CH3-C≡CH + CH3-CO-CH3 → CH3-C≡C-C(OH)(CH3)23. Конденсация алкинов со спиртами.Р. нуклеофильного присоед. спиртов к алкинам в присутствии щелочей с образованием алкенильных эфиров: CH3-C≡CH + CH3CH2OH → CH3-C(OC2H5)=CH2 Реакция Фаворского — Реппе В 1925 г. Реппе разработал пром. способ присоед-я ацетиле-на к формальдегиду. При выс. давл. в присутствии ацетиле-нида Сu происх.присоединение ацетилена к формальдегиду с обр. пропаргилового спирта и бутин-2-диола-1,4:HC≡CH + CH2O → HC≡C-CH2OH -\\\- HC≡C-CH2OH + CH2O → HOCH2C≡C-CH2OH
22.Методы создания бензольного кольца. Дегидратация и дегидроциклизация. Циклотримеризация1)Дегидроциклизацией алканов с числом ат-в С ˃ 6:---- C6H14 t, kat→ C6H6 + 4H2; 2)Сплавление солей бензойной кислоты со щелочами:
бензоат натрия
3)Реакции дегидрирования (образуются ароматич. УВ);
+ 3H2
;;
4)Циклотримеризация
Пути хим. превращений бензольного ядра:I. Реакции замещения1.Галогенирование (с Cl2, Вr2)
хлорбензол
2. Нитрование нитробензол
3. Сульфирование Бензолсульфок-та (сульфобензол):
4. Алкилир-е (образуются гомологи бензола) — реакции Фриделя-Крафтса. Алкилирование бензола происх.также при его взаимод. с алкенами:
Дегидрированием этилбензола получают стирол (винилбензол):
II. Р.присоед.1.Гидрирование циклогексан
2.Хлорирование гексахлорциклогексан (гексахлоран)
III. Р.окисл.1. Горение 2C6H6 + 15O2 12CO2 + 6H2O
Алкилирование бензола.Реакция Вюрца Фиттига
ArHal + СH3(CH2)nCH2Hal + 2Na ® Ar-CH2(CH2)nCH3 + 2NaHal Относится к реакциям кросс-сочетания. Наил.результаты достиг. при исп.арилбромидов и первичных алкилбромидов.
23. Хим. св-ва аренов. Р-и электрофильного замещения в молекуле бензола. I.Реакции замещения
1.Взаимодействие с галогенами
2.Взаимодействие с галогензамещенными алканами
3.Взаимодействие с непредельными углеводородами
4.Реакция нитрования
II.Реакции присоединения
1.Присоединение водорода
2.Присоединение хлора на свету
В реакциях электрофил. замещ. атакующей частицей явл. катионы или мол-лы с вакантной орбиталью, а также мол-лы с сильно поляризованной связью, кот. разрывается гетеролитически в процессе р-и. Как пр., электрофил. ч-цы обр. в реакционной смеси. Уходящая группа отщепляется, оставляя субстрату свою пару электронов и должна быть слабой кислотой Льюиса. В р. ароматич. электрофил. замещения такой группой чаще всего служит протон.
Механизм ароматического электрофильного замещения.
Реакция нач. с обр. донорно-акцеп. комплекса между ареном и электрофилом (p-комплекс), в кот. арен выступает в роли p-основания. Далее p-комплекс перегруппировывается в s-комплекс, содержащий s-связь углерод-электрофил. При образовании аренониевого иона ароматическая p-с-ма нарушается и образуется незамк. сопряженная с-ма циклогексадиенильного катиона, в кот. атом углерода, образующий связь с электрофилом, нах. в состоянии sp3-гибридизации, а положит. заряд делокализован по сопряженной с-ме. Делокализация положит. заряда происх. в основном за счет орто- и пара-положений по отношению к вступающему электрофилу.
Аренониевый ион является высокореакционноспособной ч-цей, кот. может стабилизироваться либо за счет присоединения нуклеофила, как это происходит при электрофильном присоединении к алкенам, либо путем отщепления протона под действием основания – энергетич. более выгодно: приводит к восстановлению ароматической с-мы. Именно оно и реализуется при ароматическом электрофильном замещении.Для многих реакций ароматического электрофильного замещения наблюдается корреляция между устойчивостью σ-комплекса и скоростью реакции. Это означает, что скорость определяется стадией образования s-комплекса и s-комплекс является хорошей моделью переходного состояния.
При смешивании аренов с электрофилами образуются слабые p-комплексы.
24. Хим св-ва аренов.Р. электрофиль замещения. Ароматическими углеводородами (аренами) называются вещества, в молекулах которых содержится одно или несколько бензольных колец — циклических групп атомов углерода с особым характером связей. Понятие “бензольное кольцо” требует расшифровки. Для этого необходимо рассмотреть строение молекулы бензола. Первая структура бензола была предложена в 1865г. немецким ученым А. Кекуле:
Эта формула правильно отражает равноценность шести атомов углерода, однако не объясняет ряд особых свойств бензола. Например, несмотря на ненасыщенность, бензол не проявляет склонности к реакциям присоединения: он не обесцвечивает бромную воду и раствор перманганата калия, т. е. не дает типичных для непредельных соединений качественных реакций.Бензольное ядро облад высокой прочностью, чем объясняется склонность к р-ям замещ. В отличие от алканов, кот также склонны к р-ям замещ, арены хар-тся большой подвижностью атомов водорода в ядре, поэтому р-ии галогенирования, нитрования, сульфирования и др. протекают в значительно более мягких условиях,чем у алканов. Обладая подвижной шестеркой p -электронов, ароматическое ядро является удобным объектом для атаки электрофильными реагентами. Этому способствует также пространственное расположение p -электронного облака с двух сторон плоского s -скелета молекулы.Для аренов наиболее характерны реакции, протекающие по механизму электрофильного замещения, обозначаемого символом-SE Механизм электрофильного замещения можно представить следующим образом. Электрофильный реагент XY (X является электрофилом) атакует электронное облако, и за счет слабого электростатического взаимодействия образуется неустойчивый p -комплекс. Ароматическая система при этом еще не нарушается. Эта стадия протекает быстро. На второй, более медленной стадии формируется ковалентная связь между электрофилом Х и одним из атомов углерода кольца за счет двух p -электронов кольца. Этот атом углерода переходит из sр2- в sр3-гибридное состояние. Ароматичность системы при этом нарушается. Четыре оставшиеся p -электрона распределяются между пятью другими атомами углерода, и молекула бензола образует карбокатион, или s -комплекс.Нарушение ароматичности энергетически невыгодно, поэтому структура s -комплекса менее устойчива, чем ароматическая структура. Для восстановления ароматичности происходит отщепление протона от атома углерода, связанного с электрофилом (третья стадия). При этом два электрона возвращаются в p -систему и тем самым восстанавливается ароматичность:
Реакции электрофильного замещения широко используются для синтеза многих производных бензола.Галогенирование. При взаимод бензола с галогеном атом водорода ядра замещается галогеном. С6Н6+Сl2=С6Н5Сl+HCl. Нитрование. При действии на бензол нитрующей смеси атом водорода замещается нитрогруппой (нитрующая смесь – это смесь концентрированных азотной и серной кислот в соотношении 1:2 соответственно). С6Н6+HNO3=C6H5NO2 нитробензол+H2O. Сульфирование осущ конц H2SO4 или олеумом. В процессе р-ии водородный атом замещается сульфогруппой. C6H6 + H2SO4 —SO3 C6H5 – SO3Hбензосульфокислота + H2O. ) Алкилирование. Замещение атома водорода в бензольном кольце на алкильную группу (алкилирование) происходит под действием алкилгалогенидов (реакция Фриделя-Крафтса) или алкенов в присутствии катализаторов AlCl3, AlBr3, FeCl3 (кислот Льюиса).С6Н6+СН3Cl=C6H5CH3+HCl. Гидрирование. Присоединение водорода осущ только в присутствии катализаторов и при повыш t. Бензол гидрируется с образованием циклогексана, а производные бензола дают производные циклогексана.C6H6+3H2=C6H12. Галогенирование.Радикальное хлорирование. В условиях радикальных р-ий (ультрафиолетовый свет, повыш t) возможно присоединение галогенов к аромат соед-ям. При радикальном хлорировании бензола получен "гексахлоран" (средство борьбы с вредными насекомыми). C6H6+3Cl2=C6H6Cl6 .К реакции электрофильного замещения относятся реакции Фриделя-Крафтса (т.е. алкилирование и ацилирование аромат ядра в присутствии AlCl3). Взаимод с н-электрофилами. Сильн к-ты с аренами образ z-комплекс и происход образование бензониевого иона. Происходит обмен атомов Н, а в присутствии дейтерированных к-т-дейтерообмен.
25 Хим св-ва аренов. Гомологи бензола. Присоед. Ароматическими углеводородами (аренами) называются вещества, в молекулах которых содержится одно или несколько бензольных колец — циклических групп атомов углерода с особым характером связей. Понятие “бензольное кольцо” требует расшифровки. Для этого необходимо рассмотреть строение молекулы бензола. Первая структура бензола была предложена в 1865г. немецким ученым А. Кекуле: Обладая подвижной шестеркой p -электронов, ароматическое ядро является удобным объектом для атаки электрофильными реагентами. Этому способствует также пространственное расположение p -электронного облака с двух сторон плоского s -скелета молекулы. Реакции в боковой цепи. По химическим свойствам алкильные радикалы подобны алканам. Атомы водорода в них замещаются на галоген по свободно-радикальному механизму. Поэтому в отсутствие катализатора при нагревании или УФ-облучении идет радикальная реакция замещения в боковой цепи. Влияние бензольного кольца на алкильные заместители приводит к тому, что замещается всегда атом водорода у атома углерода, непосредственно связанного с бензольным кольцом (a -атома углерода). Замещение в бензольном кольце возможно только по механизму SE в присутствии катализатора АlСl3: При действии на гомологи бензола перманганата калия и других сильных окислителей боковые цепи окисляются. Какой бы сложной ни была цепь заместителя, она разрушается, за исключением a -атома углерода, который окисляется в карбоксильную группу.Гомологи бензола с одной боковой цепью дают бензойную кислоту: СЕЛЕКТИВНОСТЬ .Основная реакция изомеризации сопровождается побочными реакциями - расщепления, уплотнения и др., особенно при изомеризации пентана и гексана. Для подавления побочных реакций процесс ведется в присутствии небольших количеств водорода. Помимо этих основных реакций изомеризации, элкилнафтеновые углеводороды, а также перафины могут подвергаться гидрокрекингу, в результате чего появляются низкомолекулярные парафины и изонара-финн. Поскольку термодинамические расчеты показывают, что помимо основной реакции изомеризации могут протекать реакции диспропорционирования и гидрирования ароматических углеводородов, для повышения селективности процесса необходимы катализаторы, в присутствии которых побочные реакции протекают с минимальными скоростями. Например, в присутствии катализаторов, интенсифицирующих реакции диспропорционирования, в сырье целесообразно добавлять толуол. При проведении процесса под давлением водорода добавление в сырье нафтеновых углеводородов С8 может предотвратить их образование. Поскольку термодинамические расчеты показывают, что помимо основной реакции изомеризации могут протекать реакции диспро-порайонирования и гидрирования ароматических углеводородов, для повышения селективности процесса необходимы катализаторы, в присутствии которых побочные реакции протекают с минимальными скоростями. Можно также добавлять в сырье продукты превращения ароматических углеводородов, чтобы сдвинуть равновесие в сторону образования ароматических углеводородов С8, Например, в присутствии катализаторов, интенсифицирующих реакции диспропор-ционирования, в сырье целесообразно добавлять толуол. При проведении процесса под давлением водорода добавление в сырье нафте - новых углеводородов С8 может предотвратить их образование. Реакции присоединения водорода
Присоединение хлора на свету
26Получение галогеналканов. Прямое галогенирование.Радикальным галогенированием можно получать хлор- или бромалканы. Недостаток метода : образ-ие смеси различных продуктов замещения. При этом наряду с изомерными монозамещёнными в смеси также содержатся ди- и полизамещённые соединения. К тому же, напр, эквимолярная смесь Cl2 и CH4 взрывоопасна, не говоря уже о смесях алканов со фтором. Однако меняя условия процесса можно добиться приемлемых для промышленности выходов. Напр, при хлорировании алкан берут в избытке. Продукты разделяют фракционной перегонкой. Так в промышленности получают метиленхлорид и тетрахлоруглерод.Алканы хлорируются при интенсивном УФ-облучении или нагревании. Наиболее легко образуются третичные радикалы и, соотв-но, галогеналканы; наименее – первичные. Бромирование мало характерно для алканов легче гексана, а прочие бромируются при одновременном освещении и кипячении.Или так написать CnH2n+2 + X2 CnH2n+1X + HX CnH2nX2 + HX ... В присутствии специальных реагентов, а также инициаторов свободных радикалов замещённые алкены бромируются в аллильное положение. или так писать Аллильное хлорирование осуществляется лишь при 400–600°C, обычно же идут конкурирующие реакции – присоединение по двойной связи, полимеризация, изомеризация алкенов. В промышленности так производят аллилхлорид.Замещённые арены хлорируются и бромируются в боковую цепь. Например, толуол при нагревании и интенсивном освещении хлорируют, получая бензилхлорид.Радикальным методом получают также и перфторалканы. Реакция протекает очень энергично, и вследствие большого тепловыделения приходится разбавлять фтор азотом, применяют также медные сетки для отвода теплоты. В процессе применяют переносчики фтора – фториды металлов, как CoF2, MnF2, AgF, образующие в ходе реакции соответственно CoF3, MnF4, AgF2.Получение моногалогеналканов Присоединение галогеноводородов к алкенам. R-CH=CH2+HCl→R-CHCl-CH3. Весьма легко присоединяется фтор, иод присоединяется медленно. Обычно происходит стереоселективно, кроме как в присутствии свободных радикалов. В присутствии других нуклеофилов возможно сопряжённое присоединение.Реакции спиртов с галогеноводородами. R-OH+H-Cl→R-Cl+H2O. Взаимодействие галогенидов фосфора или тионилхлорида со спиртами. 3R-OH+PCl3→3R-Cl+H3PO3. Реакция Бородина — Хунсдикера
Реакция Сварта R-Cl+AgF→R-F+AgCl. Получение дигалогеналканов Присоединение галогеноводородов к алкинам. R-C≡CH+2HCl→R-CCl2-CH3. Взаимодействие альдегидов и кетонов с PCl5, PBr5 или SF4. Рекция идёт при нагревании. R—CHO + PCl5 → R—CHCl2, Присоединение галогенов к алкенам R-CH=CH2 + Cl2 → R-CHCl-CHCl Раскрытие циклических простых эфиров при реакции с NaI в среде H3PO4+P2O5. C4H8O + HI → I-CH2CH2CH2CH2-I При 180 °C ТГФ с хлороводородом даёт 1,4-дихлорбутан. Получение галогеналкенов. С алкинами напрямую — как H-электрофилы галогеноводороды взаимодействуют медленно. Реакцию значительно ускоряют соли меди и ртути; по сути, происходит нуклеофильное присоединение галогенид-иона. Например, есть такой способ производства винилхлорида в газовой фазе на активированном угле и сулеме при 120—180 °C.Также и галогены присоединяются по тройной связи медленнее, чем по двойной. Вероятно, реакция идёт через КПЗ, который опять-таки является уже электрофильной частицй.Или применяют производные алкенов. В частности, для получения того же винилхлорида есть такие два способа.Дегидратация этиленхлоргидрина на катализаторе.Дегидрохлорирование дихлорэтана в газовой фазе при 400—500 °C. Получение галогенаренов Галогенирование аренов или алкиларенов в ядро Ph-H+Cl2→Ph-Cl Разложение арендиазониевых солей Ph-N≡N Cl→Ph-Cl Получение бензилгалогенидов Галогенирование алкиларенов в боковую цепь. Ph-CH3+Cl2→Ph-CH2Cl Хлорметилирование(р-ция Блана) Ph-H+HCHO+HCl→Ph-CH2Cl
27Хими св-ва галогеналканов. Взаимодействие галогеналканов с металлами.Взаимодействие с металлами 1--Реакция Вюрца (взаимодействие галогенпроизводных алканов с Na) CH3Cl + 2Na + ClCH3 -> 2NaCl + CH3 - CH3 -\\- 2C2H5Cl + 2Na -> 2 Nacl + C4H10 ------ CH3Cl + 2Na + C2H5Cl -> 2 NaCl + C3H8 -----------2--Гидролиз р-ва Гриньяра H2O -----RX+Mg→RMgX→RH. Р-в Гриньяра Пример Mg H2O
Бромистый втор-бутил Втор-бутил магний бромид н-бутан. 3--Восстановление металлом в кислоте RX+Zn+H+→RH+Zn+X−
Бромистый втор-бутил н-бутил. 1. Галогеналканы легко вступают в реакции замещения. Это связано с тем, что связь С-Г полярна и на атоме углерода имеется частичный положительный заряд (+). Поэтому атом углерода легко атакуется нуклеофилами, такими, как анионы ОН- и CN-, или соединениями с неподеленными электронными парами: NH3 и Н2О:. Галогенид-ион является, как говорят, «хорошей уходящей группой», поэтому реакции замещения идут достаточно легко: Примерами таких реакций служит превращение галогеналканов в спирты и амины:
Условия, необходимые для протекания таких реакции, сильно зависят от строения галогеналкана и природы нуклеофила. В некоторых случаях достаточно простого смешения реагентов, в других — требуется длительное нагревание. Легкость замещения атома галогена в значительной степени зависит и от природы галогена. Энергия связи ОТ увеличивается в ряду С—I<С—Br<С—Сl<С—F. В этом же ряду уменьшается реакционная способность галогеналканов. Связь С—F настолько прочна, что фторалканы практически не вступают в реакции нуклеофильного замещения. Активность остальных галогеналканов по отношению к нуклеофилам падает в ряду RI>RBr>RCl. 2. Галогеналканы также легко вступают в реакции отщепления. При этом образуются галогеноводород и алкен. Эти реакции протекают при действии основания на галогеналкан. Таким образом, при действии сильных оснований на Галогеналканы отщепляются молекулы НГ и образуются алкены: RСН2СН2Г+ОН-RCH2CH2OH+Г- — замещение RСН2СН2Г+ОН-RCH=CH2+Н2O+Г- — отщепление (СН3)3ССl+ОН-(СН3)2C-СН2+Н2O+Сl-
Скорости замещения и отщепления зависят также от структуры углеводородного радикала. Роль реакции замещения возрастает в следующем ряду галогеналканов: Первичные < Вторичные < Третичные. Третичные Галогеналканы вступают в реакцию замещения особенно легко (в отсутствие сильного основания):. (СН3)3ССl+Н2Oводный этанол ,25° (СН3)3СОН+НСl В целом в присутствии водного раствора основания для первичных галогеналканов характерны в основном реакции отщепления, вторичные Галогеналканы дают, как правило, смесь продуктов отщепления и замещения, а третичные образуют главным образом продукты замещения. Галогеналканы вступают в SN-реакции с различными нуклеофилами, такими, как цианид-анион CN-, ацетат-анион СН3СОO-, аммиак :NH3, амины RN..H2 и многие другие. 3. При добавлении раствора галогеналкана в диэтиловом эфире СН3СН2ОСН2СН3 к магниевой стружке происходит экзотермическая реакция: магний переходит в раствор и образуется реактив Гриньяра формулы R—Mg—Г, где R — алкильная или арильная группа, а Г — галоген. RГ+Mgэфир R-Mg-Г. Реактивы Гриньяра вступают в реакции со многими соединениями, что позволяет использовать их в синтезе самых различных веществ. Синтез реактива Гриньяра R-X+Mg→RMgX Гриньяром было показано, что в присутствии абсолютного эфира галоидопроизводные углеводородов как жирного, так и ароматического ряда реагируют с металлическим магнием, образуя галоидомагнийорганические соединения (обычно называемые просто магнийорганическими соединениями):R-Hal + Mg R-MgHal Эти соединения отличаются большой химической активностью и способны вступать в разнообразные реакции. Водой магнийорганические соединения разлагаются с образованием соответствующих углеводородов:R-MgHal + H2O R-H + Mg(OH)Hal. Реакции магнийорганических соединений играют исключительно большую роль в органической химии, так как они могут быть использованы для синтеза самых разнообразных соединений.
28.Хим сво-ва галогеналканов. Нуклеофил замещение (SNu). Механизм реакций замещения SNu1 и SNu2 1. Галогеналканы легко вступают в реакции замещения. Это связано с тем, что связь С-Г полярна и на атоме углерода имеется частичный положительный заряд (+). Поэтому атом углерода легко атакуется нуклеофилами, такими, как анионы ОН- и CN-, или соединениями с неподеленными электронными парами: NH3 и Н2О:Примерами таких реакций служит превращение галогеналканов в спирты и амины: Условия, необходимые для протекания таких реакции, сильно зависят от строения галогеналкана и природы нуклеофила. В некоторых случаях достаточно простого смешения реагентов, в других — требуется длительное нагревание. Легкость замещения атома галогена в значительной степени зависит и от природы галогена. Энергия связи ОТ увеличивается в ряду С—I<С—Br<С—Сl<С—F. В этом же ряду уменьшается реакционная способность галогеналканов. Связь С—F настолько прочна, что фторалканы практически не вступают в реакции нуклеофильного замещения. Активность остальных галогеналканов по отношению к нуклеофилам падает в ряду RI>RBr>RCl. 2. Галогеналканы также легко вступают в реакции отщепления. При этом образуются галогеноводород и алкен. Эти реакции протекают при действии основания на галогеналкан. RСН2СН2Г+ОН-RCH2CH2OH+Г- — замещение RСН2СН2Г+ОН-RCH=CH2+Н2O+Г- — отщепление (СН3)3ССl+ОН-(СН3)2C-СН2+Н2O+Сl- Скорости замещения и отщепления зависят также от структуры углеводородного радикала. Роль реакции замещения возрастает в следующем ряду галогеналканов: Первичные < Вторичные < Третичные Третичные Галогеналканы вступают в реакцию замещения особенно легко (в отсутствие сильного основания): (СН3)3ССl+Н2Oводный этанол ,25°(СН3)3СОН+НСl В целом в присутствии водного раствора основания для первичных галогеналканов характерны в основном реакции отщепления, вторичные Галогеналканы - отщепления и замещения, а третичные образуют главным образом продукты замещения. Галогеналканы вступают в SN-реакции с различными нуклеофилами, такими, как цианид-анион CN-, ацетат-анион СН3СОO-, аммиак :NH3, амины RN..H2 и многие другие. 3. При добавлении раствора галогеналкана в диэтиловом эфире СН3СН2ОСН2СН3 к магниевой стружке происходит экзотермическая реакция: магний переходит в раствор и образуется реактив Гриньяра формулы R—Mg—Г, где R — алкильная или арильная группа, а Г — галоген. RГ+Mgэфир R-Mg-Г Реактивы Гриньяра вступают в реакции со многими соединениями, что позволяет использовать их в синтезе самых различных веществ. Реакции нуклеофильного замещения — реакции замещения, в которых атаку осуществляетнуклеофил— реагент, несущий неподеленную электронную пару. Уходящая группа в реакциях нуклеофильного замещения называетсянуклеофуг. Все нуклеофилы являются основаниями Льюиса.Общий вид реакций нуклеофильного замещения: R−X + Y− → R−Y + X− (анионный нуклеофил)R−X + Y−Z → R−Y + X−Z (нейтральный нуклеофил) Выделяют реакции алифатического (широко распространены) и ароматического (мало распространены) нуклеофильного замещения. Реакции алифатического нуклеофильного замещения играют крайне важную роль в органическом синтезеи широко используются как в лабораторной практике, так и промышленности.Реакции SN1 Механизм реакции SN1 или реакции мономолекулярного нуклеофильного замещения (англ.substitution nucleophilic unimolecular) включает следующие стадии: 1. Ионизация субстратас образованиемкарбкатиона(медленная стадия): R−X → R+ + X− 2. Нуклеофильная атака карбкатиона (быстрая стадия): R+ + Y− → R−Y или (если в качестве нуклеофила выступает нейтральная частица): R+ + Y−Z → R−Y+−Z 3. Отщепление катиона(быстрая стадия): R−Y+−Z → R−Y + Z+ Примером реакции SN1 является гидролизтрет-бутилбромида: Скорость реакцииSN1 (в упрощённом виде) не зависит от концентрации нуклеофила и прямо пропорциональна концентрации субстрата[4]: Скорость реакции = k × [RX] Так как в процессе реакции образуется карбкатион, его атака (в идеальных условиях без учёта фактора влияния заместителей) нуклеофилом может происходить с обеих сторон, что приводит крацемизацииобразующегося продукта. Важно иметь в виду, чтоSN1 механизм реализуется только в случае относительной устойчивости промежуточного карбкатиона, поэтому по такому пути, обычно, реагируют только третичные ((R)3C-X) и вторичные ((R)2CH-X) алкилпроизводные. Реакции SN2 Механизм реакции SN2 или реакции бимолекулярного нуклеофильного замещения (англ.substitution nucleophilic bimolecular ) происходит в одну стадию, без промежуточного образования интермедиата. При этом атака нуклеофила и отщепление уходящей группы происходит одновременно: R−X + Y− → [Y⋯R⋯X]− → R−Y + X− Примером реакции SN2 является гидролизэтилбромида:Условный энергетический профиль реакции бимолекулярного нуклеофильного замещения представлен на диаграммеСкорость реакцииSN2 зависит как от концентрации нуклеофила, так и концентрации субстратаСкорость реакции = k × [RX] × [Y] Так как в процессе реакции атака нуклеофилом может происходить только с одной стороны, результатом реакции является стереохимическая инверсия образующегося продукта.