

29. Хим св-ва галогеналканов. Реакция элеменирования. Механизм еNu1 и еNu2. Правило Зайцева. Химические свойства галогеналканов
1. Галогеналканы легко вступают в реакции замещения. Это связано с тем, что связь С-Г полярна и на атоме углерода имеется частичный положительный заряд (+). Поэтому атом углерода легко атакуется нуклеофилами, такими, как анионы ОН- и CN-, или соединениями с неподеленными электронными парами: NH3 и Н2О:Примерами таких реакций служит превращение галогеналканов в спирты и амины:Условия, необходимые для протекания таких реакции, сильно зависят от строения галогеналкана и природы нуклеофила. В некоторых случаях достаточно простого смешения реагентов, в других — требуется длительное нагревание. Легкость замещения атома галогена в значительной степени зависит и от природы галогена. Энергия связи ОТ увеличивается в ряду С—I<С—Br<С—Сl<С—F. В этом же ряду уменьшается реакционная способность галогеналканов. Связь С—F настолько прочна, что фторалканы практически не вступают в реакции нуклеофильного замещения. Активность остальных галогеналканов по отношению к нуклеофилам падает в ряду RI>RBr>RCl.2. Галогеналканы также легко вступают в реакции отщепления. При этом образуются галогеноводород и алкен. Эти реакции протекают при действии основания на галогеналкан. RСН2СН2Г+ОН-RCH2CH2OH+Г- — замещение RСН2СН2Г+ОН-RCH=CH2+Н2O+Г- — отщепление (СН3)3ССl+ОН-(СН3)2C-СН2+Н2O+Сl- Скорости замещения и отщепления зависят также от структуры углеводородного радикала. Роль реакции замещения возрастает в следующем ряду галогеналканов: Первичные < Вторичные < Третичные Третичные Галогеналканы вступают в реакцию замещения особенно легко (в отсутствие сильного основания): (СН3)3ССl+Н2Oводный этанол ,25°(СН3)3СОН+НСl В целом в присутствии водного раствора основания для первичных галогеналканов характерны в основном реакции отщепления, вторичные Галогеналканы - отщепления и замещения, а третичные образуют главным образом продукты замещения. Галогеналканы вступают в SN-реакции с различными нуклеофилами, такими, как цианид-анион CN-, ацетат-анион СН3СОO-, аммиак :NH3, амины RN..H2 и многие другие. 3. При добавлении раствора галогеналкана в диэтиловом эфире СН3СН2ОСН2СН3 к магниевой стружке происходит экзотермическая реакция: магний переходит в раствор и образуется реактив Гриньяра формулы R—Mg—Г, где R — алкильная или арильная группа, а Г — галоген. RГ+Mgэфир R-Mg-Г Реактивы Гриньяра вступают в реакции со многими соединениями, что позволяет использовать их в синтезе самых различных веществ. Элиминирование (от лат.elimino — изгоняю) — это отщепление двух атомов или групп атомов от соседних атомов углерода с образованием между ними π-связи. Атомы углерода при элиминировании переходят из sр3- в sp2-гибридное состояние (или из sр2- в sp).Реакция элиминирования может проходит в одну стадию (по механизму E2), либо в две стадии, по механизму E1 Исходными веществами могут служить представители разных классов органических соединений.Зайцева Правило : при дегидратации вторичных и третичных спиртов в присут. сильных к-т и при дегидрогалогенировании вторичных и третичных алкилгалогенидов под действием оснований протон отщепляется преим. от наименее гидрогенизир. атома С с образованием соед. II:Элиминирование по 3. п. протекает в ряде случаев при разложении вторичных и третичных солей диазония (X = N2+), а также вторичных и третичных алкил- и арилсульфонатов (X = AlkSO2O, ArSO2O) под действием оснований. В практике орг. синтеза обычно не встречаются примеры р-ций элиминирования, в к-рых 3. п. выполнялось бы абсолютно; реально получаются смеси изомерных олефинов ф-л II и III. При элиминировании НХ из соед. I получающийся наиб. замещенный олефин (ф-лы II) наз. образующимся по 3. п., а терминальный олефин III – образующимся по правилу Гофмана (см. Гофмана реакции ). Элиминированию по 3. п. способствует наличие в соед. I легко удаляющейся группы X; способность к такому отщеплению уменьшается в ряду N2+ > I- > Br- > Cl- > TsO- > R2S+ > F- > R3N+ (Ts – CH3C6H4SO2, R – алифатич. радикал). Напр., при обработке 2-бром-, 2-тозилокси- и 2-диметилсульфониобутана этилатом Na в этаноле соотношения образующихся 2-бутена и 1-бутена составляют соотв. 81:19, 65:35 и 26:74. В р-циях, протекающих под действием алкоголятов, элиминированию по 3. п. благоприятствует применение оснований с радикалом небольшого объема. Так, при использовании С 2 Н 5 ОК, трет-C4H9OK, трет -С 5 Н 11 ОК и (С 2 Н 5)3 СОК в р-ции дегидрогалогенирования 2-метил-2-бромбутана соотношения образующихся соед. IV и V составляют соотв. 62:38, 27:73, 22:78 и 11:89: Увеличение объема радикалов R и R’ в исходном соед. препятствует протеканию р-ции по 3. п., напр.: Образованию олефинов по 3. п. способствует применение биполярных протонных р-рителей. Так, при проведении р-ции в этаноле и ДМСО соотношения продуктов VI и VII составляют соотв. 65:35 и 46:54. Правило сформулировано А. М. Зайцевым в 1875.
30.Классиф гидроксилпроизводных углеродов, номенклатура.Изомерия.Производные углеводородов, в молекулах которых один или несколько атомов водорода замещены гидроксильными группами (ОН), называют предельными спиртами или алкоголями. Общая формула R-OH. Спирты классифицируются: 1) по строению углеводородного радикала различают: а) спирты алифатического (жирного ряда), Аlk-ОН; б) ароматические, которые разделяются на фенолы Аr-OH и жирноароматические спирты Ar(CH2)n-OH; 2) по числу гидроксилов спирты бывают одно-, двух– и многоатомные. Например: а) одноатомные спирты СН3-ОН (метанол); б) двухатомный спирт HO-CH2-CH2OH (этандиол); в) трехатомный спирт НОСН2-СНОН-СН2ОН (глицерин). В зависимости от характера углеродного атома, при котором находится гидроксил, различают первичные, вторичные и третичные спирты. 1) R-CH2-OH, или Аr-СН2-ОН, – первичный спирт; 2) – вторичный спирт; 3) – третичный спирт. Изомерия и номенклатура. Изомерия спиртов зависит от строения углеводородной цепи и положения гидроксила в цепи. Спирты часто называют по радикально-спиртовой и систематической (ИЮПАК) номенклатуре. При названии спирта по радикально-спиртовой номенклатуре в основе лежит название соответствующего углеводородного радикала, связанного с гидроксилом, с прибавлением окончания – овый спирт. Например: 1) СН3-ОН – метиловый спирт (древесный); 2) С2Н5-ОН – этиловый спирт; 3) н-С4Н9ОН – бутиловый спирт; 4)
– трет-бутиловый спирт. В основе названия спирта по ИЮПАК лежит наименование углеводорода самой длинной углеводородной цепи, наличие же гидроксильной группы указывается окончанием – ол, с цифрой за ним, указывающей номер атома углерода, при котором стоит гидроксил. При этом углеродная цепь нумеруются таким образом, чтобы гидроксил имел наименьший номер. Заместительная номенклатура используется достаточно редко. По ней спирты называют как производные карбинола (метанола). Например, фенилкарбинол – бензиловый спирт (оксиметилбензол), этилкарбинол – пропиловый спирт (пропанол), винилкарбинол – пропен-2-ол-1 (аллиловый спирт) .Тривиальная номенклатура, напротив, до сих пор широко применяется. Метанол (метиловый спирт, муравьиный спирт), пропиловый спирт, группа амиловых спиртов (С5) и т.д. Не говоря уже о непредельных спиртах, которые практически только и называются тривиальными названиями – аллиловый и пропаргиловый спирты. Изомерия. Изомерия спиртов аналогична изомерии галогенопроизводных. В случае спиртов кроме изменения строения углеродного скелета может изменяться положение –OH группы.
31 Способы получ спиртов.Гидролизгалогеналканов. Гидратация алкенов. Наиболее общим способом является получение спиртов из галоидных соединений обменом атомат галоида на гидроксил: Этим способом можно получать спирты первичные, вторичные и третичные в зависимости от строения радикала СnН2n+1 Иногда для получения спирта из галоидного алкила сначала получают сложный эфир уксусной кислоты взаимодействием галоидного алкила с уксуснокислым серебром, например
а затем омылением сложного эфира получают спирт:
Гидролиза галогеналканов
Гидратация алкенов:CH2=CH2 + H2O (кат.) CH3CH2OH Присоединение воды к несимметричным алкенам идет по правилу Марковникова с образованием вторичных и третичных спиртов CH3–CH=CH2 +H2O(кат.) CH3CH(OH)CH3 ---\\\--- (CH3)2C=CH2 + H2O (кат.) (CH3)3C–OH
Восстановление Альдегиды и кетоны способны восстанавливаться, т. е. присоединять атомы водорода, причем главными продуктами восстановления являются спирты: первичные — из альдегидов и вторичные — из кетонов.Восстановление кетонов часто может идти также и в другом направлении, а именно с присоединением атома водорода к кислородукарбонильной группы, причем атомы углерода карбонильных групп двух молекул связываются между собой, например:
Таким образом получаются двухатомные спирты с удвоенным числом атомов углерода в молекуле, сравнительно с молекулойисходного кетона, — так называемые пинаконы.Возможно и более глубокое восстановление карбонильной группы — замещение атома кислорода двумя водородными атомами(элиминирование карбонильной группы).Взаимодействием реактива Гриньяра с формальдегидом можно получить практически любой первичный спирт (кроме метанола). Для этого продукт присоединения реактива Гриньяра гидролизуют водой:
Н2O Н2СО + RMgX → R-CH2-O-MgX → R-CH2-OH . -Mg(OH)X Оксосинтез является одним из важнейших нефтехимических процессов, с помощью которого производится широкий ассортимент кислородсодержащих соединений - альдегидов, спиртов, кислот, сложных эфиров, кетонов и др. Эти продукты могут быть получены на основе доступных дешевых источников сырья - газов крекинга и пиролиза, продуктов дегидрирования парафинов, жидких продуктов термической переработки нефти, полимер-олефинов.Реакция гидроформилирования основана на взаимодействий олефинов с окисью углерода и водородом. Катализаторами реакции являются карбонилы кобальта, а также карбонилы других металлов VIII группы таблицы Менделеева.Наиболее признанный механизм:
32Хим св-ва спиртов.Р. с сохран. Атома кислорода. Алкоголята. Спирты - органические соединения, содержащие одну или более гидроксильных групп(гидроксил, −OH), непосредственно связанных с насыщенным (находящемся в состоянии sp³ гибридизации) атомом углерода[1]. Спирты можно рассматривать как производные воды (H−O−H), в которых один атом водорода замещен на органическуюфункциональную группу: R−O−H.В номенклатуре IUPAC для соединений, в которых гидроксильная группа связана с ненасыщенным находящемся в состоянии sp2гибридизации атомом углерода, рекомендуются названия «енолы» (гидроксил связан с винильной C=C связью)[2] и «фенолы» (гидроксил связан с бензольным или другим ароматическим циклом).Реакция альдегидов со спиртами. Синтез полуацеталей и ацеталей. В благоприятных условиях (например: а) при нагревании с кислотой или в присутствии водоотнимающих средств; б) при внутримолекулярной конденсации с образованием пяти- и шестичленных циклов) альдегиды реагируют со спиртами. При этом к одной молекуле альдегида может присоединиться либо одна молекула спирта (продукт – полуацеталь), либо две молекулы спирта (продукт – ацеталь):
Кисло́ты — сложные вещества, в состав которых обычно входят атомы водорода, способные замещаться на атомы металлов, икислотный остаток. Водные растворы кислот имеют кислый вкус, обладают раздражающим действием, способны менять окраскуиндикаторов, отличаются рядом общих химических свойств. АЛКОГОЛЯТЫ, продукты замещения атома Н в молекуле спирта на металл (М). Алкоголяты одноатомных спиртов. Их общая ф-ла M(OR)n, где n-степень окисления металла. Алкоголяты щелочных, щел.-зем.металлов, Т1(1) и первичных спиртов-ионные соед.; неплавки, нелетучи; т. разл. 200-300 °С; раств. в спиртах и жидком NH3;электролиты в р-ре. Из спиртовых р-ров обычно выделяются в виде кристаллосольватов. Производные металлов III-VIII групп и спиртов(начиная с С2Н5ОН), а также M1OR-mpem- молекулярные мономерные или олигомерные соед.; имеют низкие т-ры плавления и кипения; хорошо раств. в неполярных р-рителях, плохо-в спиртах; р-ры не проводят ток. Метилаты тех же элементов-обычно координац.полимеры; неплавки, нелетучи; не раств. ни в одном из р-рителей. Большинство алкоголятов элементов середины периодич. системы сочетают св-ва ионных и молекулярных соед. Все алкоголяты очень гигроскопичны. Образование алкоголятов Водородный атом гидроксильной группы при действии щелочных металлов способен заменяться наатомы этих металлов, причем получаются твердые, растворимые в спирте соединения, называемые алкоголятами:
Первичные спирты при окислении образуют альдегиды, которые затем легко окисляются до карбоновых кислот.
При окислении вторичных спиртов образуются кетоны.
Третичные спирты более устойчивы к действию окислителей. Они окисляются только в жестких условиях (кислая среда, повышенная температура), что приводит к разрушению углеродного скелета молекулы и образованию смеси продуктов (карбоновых кислот и кетонов с меньшей молекулярной массой).Процесс идет через стадию дегидратации спирта с последующим деструктивным (жестким) окислением алкена. Например:
33Хим св-ва спиртов. Нуклеофил замещ гидроксо группы. Взаимодействие спиртов с галогенводородными кислотами. Спирты - органические соединения, содержащие одну или более гидроксильных групп(гидроксил, −OH), непосредственно связанных с насыщенным (находящемся в состоянии sp³ гибридизации) атомом углерода[1]. Спирты можно рассматривать как производные воды (H−O−H), в которых один атом водорода замещен на органическуюфункциональную группу: R−O−H.В номенклатуре IUPAC для соединений, в которых гидроксильная группа связана с ненасыщенным находящемся в состоянии sp2гибридизации атомом углерода, рекомендуются названия «енолы» (гидроксил связан с винильной C=C связью)[2] и «фенолы» (гидроксил связан с бензольным или другим ароматическим циклом)Реакции нуклеофильного замещения ОН группы Для проведения SN-реакций в спиртах необходимо предварительно модифицировать ОН группу, превратив ее в хорошую уходящую группу. Для этого спирты переводят в оксониевые соединения или эфиры неорганических кислот:
В биохимических процессах замещение ОН группы протекает через стадию превращения спиртов в эфиры фосфорной, дифосфорной и трифосфорной кислот, анионы которых являются чрезвычайно легко уходящими группами: Замещение ОН группы на галоген Спирты взаимодействуют с галогеноводородами и галогенидами фосфора и серы с образованием галогенпроизводных. 1. Действие галогеноводородов. R-OH + HHal ® R-Hal + H2O Реакция протекает через стадию протонирования ОН группы с последующим нуклеофильным замещением по механизму SN1 илиSN2. Первичные спирты реагируют по механизму SN2. Реакция идет медленно, так как в протонных растворителях (вода, спирты) галогенид-ионы – слабые нуклеофилы. Вторичные и третичные спирты реагируют по механизму SN1. Таким образом, реакционная способность спиртов возрастает в ряду: первичный, вторичные, третичные Реакционная способность галогеноводородов увеличивается в ряду: HCl параллельно с возрастанием силы кислот и нуклеофильности галогенид-анионов. Однако HI не может быть использована, так как легко восстанавливает спирты до углеводородов. HBr реагирует с первичными, вторичными и третичными спиртами. HCl действует только на третичные спирты.
Для вторичных и первичных спиртов требуется присутствие катализатора – кислоты Льюиса.
34Хим св-ва спиртов. Взаимодействие спиртов с серной кислотой: Образование простых эфиров При нагревании спиртов в присутствии серной кислоты происходит замещение ОН группы на группу OR и образуются простые эфиры Реакция протекает через стадию протонирования спирта с последующим нуклеофильным замещением, в котором одна молекула спирта (протонированная) выполняет роль субстрата, а вторая – нуклеофила. В отличие от спиртов, являющихся слабыми нуклеофилами, алкоголяты, образующие алкоксид-ионы RO− — сильные нуклеофилы и легко реагируют с алкилгалогенидами по механизму SN2 с образованием простых эфиров: Вместо алкилгалогенидов можно использовать также алкилсульфонаты Побочными продуктами реакции являются алкены, образующиеся в результате конкурирующего процесса элиминирования спирта:
Межмолекулярная и внутримолекулярная дегидратация спиртовПри осторожном нагревании в присутствии серной кислоты происходит межмолекулярная дегидратация спиртов с образованием простых эфиров. Если в реакцию с кислотой вступают двухатомные спирты, будет протекать реакция внутримолекулярной дегидратации с образованием гетероциклических соединений. Так например, 1,4-бутандиол образует тетрагидрофуран Так как реакция получения эфира обратима, для её смещения вправо обычно используют метод отгонки конечных продуктов (воды или эфира) из реакционной смеси. Существуют и методы термокаталитической дегидратации спиртов. Например, первичные спирты в присутствии смешанного Ni−Al2O3−SiO2 катализатора и водорода при нагревании превращаются в простые эфиры:
Метод межмолекулярной дегидратации — один из наиболее старых способов получения эфиров — используется весьма ограниченно и только для неразветвлённых первичных спиртов из-за высокой доли алкенов, образующихся в случае внутримолекулярной дегид5ратации при использовании вторичных и третичных спиртов. Спирты способны образовывать сложные эфирыв реакциях сорганическими кислотамипри нагревании в присутствии кислотногокатализатора(как правило, концентрированнойH2SO4). Этот процесс получил название кислотно-каталитической реакции этерификации(также известен какреакция Фишера). Например, взаимодействиеэтаноласуксусной кислотойдаетэтилацетат:
Механизм реакции:
Кислотно-каталитическая реакция этерификации — простейший и наиболее удобный метод получения сложных эфиров для случая, когда ни кислота, ни спирт не содержат чувствительных функциональных групп. В качестве катализатора, помимо традиционно используемой серной кислоты, могут выступать кислота ЛьюисаилиБренстеда; растворителем, обычно, служит сам спирт или, если это невозможно —толуолиликсилол. Для увеличения выхода эфира используют отгонку или химическое связывание воды, а также специализированное лабораторное оборудование —аппарат Дина — Старка. Реакция между спиртом и кислотой происходитвприсутствии дициклогексилкарбодиимида(ДЦК)и небольших количеств 4-N,N-диметиламинопирнидина. ДЦК и карбоновая кислота на первом этапе образует O-ацилизомочевинный интермедиат, который в дальнейшем вступает в реакцию со спиртом, образуя сложный эфир:
35.Функц. карбонильные группы. Классиф. карбон. соединений.Гомолог. ряды альдегидов и кетонов. Ном-тура и изомерия. Р-ции окисл. алкенов; ок-ия и дегидрирования алканолов; гидролиз дигалогеналкан гидратация алкинов; оксосинтез; пиролиз кальциевых солей карбоновых кислот, пинаколиновая перегруппировка. Карбонильные соединения содержат в молекуле карбонильную группу Карбонильные соединения делятся на альдегиды и кетоны. Свойства альдегидов и кетонов определяются строением карбонильной группы C=O. Систематические названия альдегидов строят по названию соответствующего углеводорода и добавлением суффикса -аль. Нумерацию цепи начинают с карбонильного атома углерода. Тривиальные названия производят от тривиальных названий тех кислот, в которые альдегиды превращаются при окислении. Названия альдегидов по заместительной номенклатуре в соответствии с правилами ИЮПАК производят из названия соответствующего углеводорода с добавлением окончания -аль. Перед корнем названия записывают боковые заместители с указанием их положения их числа. Нумерация атомов углерода начинается с углеродного атома карбонильной группы. Получение: 1. Гидратация алкинов. Hg2+, H+ ---\\\--- СH≡СН + Н2О → СН3-СН=О.2. Общий способ получения карбонильных соединений окисление спиртов . СН3-СН2-ОН + СuО t→ СН3-СН=О + Сu + Н2О .3. При щелочном гидролизе дигалогеналканов, содержащих два атома галогена при одном атоме углерода, образуются двухатомные спирты, в которых две группы ОН соединены с одним атомом углерода. Эти вещества неустойчивы и отщепляют воду, превращаясь в карбонильные соединения:СН3-СНСl2→[СН3СН(ОН)2] 2NaOH СН3-СН=О +Н2О. 4. Дегидрирование спиртов. ZnO, 380° С . СН3-СН(ОН)-СН3 → СН3-СО-СН3
5. Окисление алкенов. Альдегиды и кетоны получают окислением углеводородов ряда этилена кислородом воздуха в присутствии хлоридов палладия (II) и меди (I), например: СH2-CH2+1/2O2 PBCL2*CUCL2 CH3CH=O Этим экономичным способом в промышленности получают низшие альдегиды и кетоны. 6. (оксосинтез), присоединение оксида углерода и водорода к олефинам в присутствии катализатора с образованием альдегидов, например СН3СН = СН2 + СО + Н2→СН3СН2СН2СНО. 7/ Пинаколиновая перегруппировка, образ-ие кетонов при действии кислот (HCI, H2SO4), а также ZnCl2 на пинаконы; при этом происходит дегидратация, сопровождающаяся изменением скелета молекулы — миграцией одного из заместителей к соседнему углеродному атому. Отход гидроксильной группы и перемещение заместителя происходят синхронно
36.Хим св альдегидов и кетонов. Реакции нуклеофильного присоединения. Альдегиды и кетоны, обладая электрофильным центром, способны вступать во взаимодействие с нуклеофильными реагентами. Для оксосоединений наиболее характерны реакции, протекающие по механизму нуклеофильного присоединения обозначаемому АN. Реакция с циановодорооной (синильной) кислотой. приводящее к образованию a- оксинитрилов. Эта реакция используется для удлинения углеродной цепи и получения a- оксикислот. СH3-СH=O+H-CN CH3-CH(CN)-OH Присоединение гидросульфитов служит для выделения альдегидов из смесей с другими веществами и для получения их в чистом виде, поскольку полученное сульфопроизводное очень легко гидролизуется:
R-CH=O+NaHSO3 R-CH-SO3Na
|
OH
Присоединение металлоорганических соединений При этой реакции углеводородный радикал металлоорганического соединения присоединяется к углероду, а остальная часть его молекулы — к кислороду, например:
CH3-CH=O+R-MGI CH3-CH-OMg
|
R
При разложении полученных соединений водой получаются соответствующие спирты.
Наряду с этой реакцией, при действии магнийорганических соединений на кетоны происходит изомеризация кетонов в ненасыщенные спирты и образование смешанных магниевых алкоголятов этих спиртов (Гриньяр):
37.Хим. св-ва альдегид. и кетонов. О-В реакции. Восстановление альдегидов и кетонов в спирты. При восстановлении альдегидов образуются первичные, а при восстановлении кетонов — вторичные спирты
O
// 2H
H3C—C H3C—CH2OH
\ H
уксусный этиловый альдегид спирт
2H
H3C—CO—CH3 H3C—CH—CH3
|
OH
ацетон изопропиловый
спирт
Одной из качественных реакций для обнаружения альдегидной группы является реакция “серебряного зеркала” — окисление альдегидов оксидом серебра.
В растворе аммиака оксид серебра образует комплексное соединение, при действии которого на альдегид происходит окислительно-восстановительная реакция. Альдегид окисляется в соответствующую кислоту (точнее, в ее аммонийную соль), а комплексный катион восстанавливается до металлического серебра, которое дает блестящий налет на стенках пробирки — “серебряное зеркало”:
Другая качественная реакция на альдегиды заключается в окислении их гидроксидом меди (II). При окислении альдегида светло-голубой гидроксид меди (II) превращается в желтый гидроксид меди (I) при комнатной температуре. Если подогреть раствор, то гидроксид меди (I) превращается в оксид меди (I) красного цвета, который плохо растворим в воде и выпадает в осадок:
При прибавлении фуксинсернистой кислоты к раствору альдегида смесь приобретает красное или красно-фиолетовое окрашивание. При последующем прибавлении минеральных кислот это окрашивание, как правило, исчезает; исключение составляет формальдегид; окрашивание фуксинсернистой кислоты, вызванное формальдегидом, не исчезает от прибавления кислот.
38.Хим св-ва альдегидов и кетонов. Конденсация. Хим св-ва обусловлены наличием в их молекулах сильно полярной карбонильной группы (связь поляризована в сторону атома кислорода). Чем больше частичный заряд(+)на атоме C этой группы, тем выше активность соединения.1.Горение: 2CH3CHO+5O2=4CO2+4Н2О 2CH3COCH3 + 9O2 6CO2 + 6H2O 2.Присоединение(по двойной связи карбонилгруппы). В ряду HCHO RCHO RCOR' склонность к реакциям присоединения уменьшается. Это связано с наличием и числом углеводородных радикалов, связанных с атомом С карбонильной группы. а) Гидрирование (восстановление водородом): HCHO + H2 CH3OH CH3—CO—CH3 + H2 CH3—CH(OH)—CH3 Из альдегидов при этом получаются первичные спирты, а из кетонов - вторичные. 3)Окисление: CH3CHO + Ag2O 2Ag + CH3COOH (р-ция "серебряного зеркала" – качеств реакция) HCHO + 2Cu(OH)2 2H2O + Cu2O + HCOOH (образуется красный осадок - качественная реакция).Кетоны слабыми окислителями не окисляются.4.Замещение атомов водорода в углеводородном радикале (замещение происходит в -положение, т. е. замещается атом водорода у 2-го атома углерода):
|
3 |
2(a) |
1 |
|
|
CH3 |
—CH2 |
—CHO |
+ Cl2 CH3—CHCl—CHO + HCl |
АЛЬДОЛЬНАЯ КОНДЕНСАЦИЯ, взаимод. Двух молекул альдегида или кетона (одинаковых или разных) в присут. к-т или оснований с образованиемгидроксиальдегидов (альдолей), напр.: Р-ция обратима и может осуществляться только при наличии хотя бы у одного реагента атома Н вположении к карбонильной группе.Кетоны реагируют значительно труднее альдегидов.
КРОТOНОВАЯ КОНДЕНСАЦИЯ, взаимодействие двух молекул альдегидов или кетонов друг с другом в присут. оснований или к-т, сопровожд отщеплением воды и образованием -непредельного карбонильного соед.Первая стадия процесса - синтез альдоля , к-рый обычно при комнатной т-ре или при нагр. дегидратируется. Кетоны реагируют в более жестких условиях, чем альдегиды. Образующееся в р-ции нeнасыщ. карбонильное соед. может вступать в р-цию с исходными в-вами, напр Механизм дегидратации альдоля зависит от природы катализатора (основания или к-ты), напр.: Кротоновую конденсацию широко используют для синтеза ненасыщ. альдегидов и кетонов (напр., для пром. получения кротонового альдегида).
39.Амины. Классиф, номенкл, изомер. Амины-производные аммиака, в молекуле которого 1,2 или 3 атома водорода замещены органическими радикалами. Радикалы могут быть алифатическими (насыщенными или ненасыщ), карбоциклическими, ароматическими или гетероциклическими. В зависимости от числа органических радикалов у атома азота амины делятся на первичные - RNH2, вторичные - R2NH и третичные – R3N. При этом не имеет значения, какие органические радикалы (первичные, вторичные или третичные) выступают в роли заместителя – в первичных, вторичных и третичных аминах могут присутствовать как первичные, так и вторичные и третичные алкильные радикалы. Существуют также четвертичные соли аммония R4N+X- - производные иона аммония, у которого все валентности атома азота заняты органическими заместителями.Для обозначения аминов используют 3вида номенклатуры – тривиальную (напр, анилин, толуидин), заместительную (метиламин, триметиламин) и номенклатуру ИЮПАК (1-аминогексан). Для обозначения простейших аминов наиб часто применяют заместительную номенклатуру, согласно которой названия аминов строятся путем перечисления углеводородных радикалов в алфавитном порядке с добавлением окончания амин. В более сложных случаях используют номенклатуру ИЮПАК, в которой амины рассматривают как производные углеводородов с префиксом амино-. В классе аминов существуют след виды изомерии: изомерия углеводородного скелета (напр, 1-аминобутан и 1-амино-2-метилпропан), положения аминогруппы (напр, 1- и 2-аминобутаны), для полизамещенных ароматических аминов – изомерия взаимного расположения заместителей (напр, орто-, мета- и пара-толуидины) и изомерия первичных, вторичных и третичных аминов (напр, н-пропиламин, метилэтиламин и триметиламин).1- Наиболее общим методом получения первичных аминов является восстановление нитросоединений: . Важнейший ароматический амин - анилин - образуется при восстановлении нитробензола (восстановители - водород в присутствии металлических катализаторов, Fe + HCl, сульфиды): Эта реакция носит имя русского химика Н.Н. Зинина, осуществившего ее впервые в 1842 г. 1) Восстановление амидов (восстановитель - алюмогидрид лития LiAH4): Восстановление нитрилов с образованием первичных аминов:R-CN + 4[H] R-CH2NH2 Этим способом в промышленности получают гексаметилендиамин, который используется в производстве полиамидного волокна найлон. Получение аминов путём введения алкильных групп в молекулы аммиака и аминов (реакции алкилирования).
При нагревании галогеналканов с аммиаком образуется смесь первичных, вторичных и третичных аминов.
В основе этих превращений лежит реакция нуклеофильного замещения галогена в галогеналканах. Роль нуклеофила играют молекулы аммиака и аминов, имеющие неподеленную пару электронов на атоме азота. Действием галогеналканов на первичные алифатические и ароматические амины получают вторичные и третичные амины.
40.Хим.св-ва аминов(обр-е солей,р-ции ацилирования и алкилирования,взаим-е с азотист.к-той) 1)Алифатические амины взаимод-т с кислотами с образ-м солей.В водных растворах соли орг.аминов полностью диссоциир-т.С₂Н₅NH₂+HCl→[C₂H₅NH₃]Cl; --- [(CH₃)₂NH₂]Cl↔[(CH₃)₂NH₂]⁺+Cl. Под действ-м щелочей соли аминов разлаг.с выделением свобод.аминов: [СН₃NH₃]Cl +NaOH→CH₃-NH₂+NaCl+H₂O Как и аммиак,орг.амины обр-т комлекс.соли:AgNO₃+2CH₃NH₂→[Ag(CH₃NH₂)₂]NO₃ Ароматич.амин-анилин при взаимод-и с кис-ми обр.соли фениламмония. C₆H₅NH₂+HCl→[C₆H₅NH₃]⁺Cl⁻ Под дейст-м щелочи соли анилина разлаг.с выд-м своб.анилина. [C₆H₅NH₃]Cl+NaOH→ C₆H₅Na+NaCl+H₂O 2)Алкилирование-ведет к превращ-ю первич.аминов во вторич.и третич. СН₃ NH₂+ CH₃I→(CH₃)₂ NH+HI; (CH₃)₂ NH+ CH₃I→(CH₃)₃N+HI; Алкилирование аминогруппы анилина спиртами или галогеналканами ведет к обр-ю алкиламинов C₆H₅NH₂+ CH₃I→ HI+ C₆H₅NH- СН₃ Ацилирование аминов хлорангидридами кислот ведет к обр-ю замещенных амидов кислот:СН₃СОСl+ СН₃ NH₂→HCl+СН₃СО-NH-СН₃(Метилацетамид) Ацилирование анилина ангидридами или хлорангидридами кислотведет к обр-ю замещ-х анилидов:СН₃СОСl+ C₆H₅NH₂→HCl++ C₆H₅NH-CO- СН₃(ацетанилид) 3)Окисление азотист.кисл-й.Первич.амины окисл.до спиртов,вторич.до нитрозаминов,а третич.не взаимод-т. СН₃-СН₂- NH₂+HNO₂→ СН₃-СН₂-OH+N₂+ H₂O -- (CH₃)₂ NH+ HNO₂→ H₂O+ (СН₃)₂-N-N=О В р-ции анилина с азотист.к-той обр.соли диазония,кот.явл. промеж.продуктами в проц-х синтеза анилиновых красителей.C₆H₅NH₂+ HNO₂+HCl→2 H₂O+[C₆H₅-NΞN]⁺Cl⁻.
41.Классиф-я монокарбоновых кислот. Гомологич.ряд,номенклатура,изомерия. Монокарб-е кислоты-это производные УВ,содерж-е в молекуле одну функц-ю карбоксил.группу-СООН.Монокарб.к-ты подразд-тв завис-ти от природыуглеводор.остатка:насыщенные;ненасыщен.;ароматические.Номенклатура:1.тривиальная,2.рациональная,3.систематическая.НСООНмуравьиная,,метановая;СН₃СООНуксусная,уксусная,этановая;СН₃СН₂СООНпропионовая,метилуксусная,пропановая,СН₃СН₂СН₂СООН-масляная,этилуксусная,бутановая.Изомерия:1. Изомерия углеродной цепи. Она начинается с бутановой кислоты (С3Н7СООН), которая существует в виде двух изомеров: масляной и изомасляной (2-метилпропановой) кислот.2. Изомерия положения кратной связи, например:СН2==СН—СН2—СООН СН3—СН==СН—СООН Бутен-3-овая кислота Бутен-2-овая кислота (винилуксусная кислота) (кротоновая кислота) 3. Цис-, транс-изомерия, например:
4. Межклассовая изомерия: например, масляной кислоте (СН3—СН2—CH2—СООН) изомерны метиловый эфир пропановой кислоты (СН3—СН2—СО—О—СНз) и этиловый эфир уксусной кислоты (СН3—СО—О —CH2—СН3). 5. У гетерофункционалъных кислот возможна изомерия, связанная с положением функциональных групп, например, существуют три изомера хлормасляной кислоты: 2-хлорбутановая, 3-хлорбутановая и 4-хлорбутановая. Для гетерофункциональных кислот возможна также оптическая изомерия.
42.Методы синтеза. Реакции окисления спиртов, альдегидов. Получение жирных кислот окислением парафинов. Окисление спиртов Окислители – KMnO4, K2Cr2O7+H2SO4, CuO, O2+катализатор. Легкость окисления спиртов уменьшается в ряду:первичные ≥ вторичные >> третичные.Первичные спирты при окислении образуют альдегиды, которые затем легко окисляются до карбоновых кислот.
При окислении вторичных спиртов образуются кетоны.
Третичные спирты более устойчивы к действию окислителей. Они окисляются только в жестких условиях (кислая среда, повышенная температура), что приводит к разрушению углеродного скелета молекулы и образованию смеси продуктов (карбоновых кислот и кетонов с меньшей молекулярной массой). Процесс идет через стадию дегидратации спиртас последующим деструктивным (жестким)окислением алкена. Например:
Предельное окисление гидроксисоединений до CO2 и Н2О происходит при их горении, например:
2CH3OH + 3O2 2CO2 + 4H2O
Полное окисление метанола идет схеме:
При сгорании спиртов выделяется большое количество тепла. C2H5OH + 3O2 2CO2 + 3H2O + 1370 кДж Благодаря высокой экзотермичности реакции горения этанола, он считается перспективным и экологически чистым заменителем бензинового топлива в двигателях внутреннего сгорания. В лабораторной практике этанол применяется как горючее для "спиртовок".Окисление альдегидов. Адьдегиды легко окисляются различными окислителями. Конечным продуктом окисления.
43.Хим свокарбоновых кислот. Кислотные свойства. Сравнение кислотных свойств.Наиболее важные химические свойства, характерные для большинства карбоновых кислот:1. Карбоновые кислоты при реакции с металлами, их оксидами или их осно́вными гидроксидами дают соли соответствующих металлов:
2. Карбоновые кислоты могут вытеснять более слабую кислоту из её соли, например:
3. Карбоновые кислоты в присутствии кислого катализатора реагируют со спиртами, образуя сложные эфиры (реакция этерификации):
4. При нагревании аммонийных солей карбоновых кислот образуются их амиды:
5. Под действием карбоновые кислоты превращаются в соответствующие хлорангидриды:
Карбоновые кислоты, имеющие Ка ˜ 10-5, как кислоты слабее таких минеральных кислот, как HCl, HBr, HF, H2SO4, HNO3, H3PO4, но намного сильнее спиртов, фенолов, угольной кислоты.
Карбоксилат-ион в отличие от алкоголят-иона стабилизирован сопряжением, что доказано рентгеноструктурным анализом (длины обеих связей С-О одинаковы).
Заместители в фрагменте R, стабилизирующие карбоксилат-ион, то есть облегчающие его образование, сильно увеличивают кислотность алифатических кислот. Такую роль играют электроноакцепторные заместители.Влияние заместителей в ароматических карбоновых кислотах менее заметно, чем в алифатических, но ожидаемое; электроноакцепторные заместители и сопряжение усиливают кислотность. Как известно, кислотно-основные взаимодействия идут с образованием более слабых кислот и оснований, чем исходные, поэтому карбоновые кислоты реагируют с оксидами, гидроксидами щелочных, щелочноземельных металлов, карбонатами, фенолятами, алкоголятами, амидами идр.
Образование карбоксилат-аниона сопровождается усилением нуклеофильности карбоновых кислот и облегчает процесс декарбоксилирования.карбоновые кислоты, реагируя с сильными кислотами, ведут себя, как слабые основания, при этом возможно протонирование как карбонильного, так и гидроксильного атома кислорода.
Следует отметить, что на легкость отщепления протона, т.е. на проявление кислотных свойств, существенное влияние оказывает природа углеводородного радикала, связанного с карбоксильной группой. Так, например, алкильные радикалы, обладающие электронодонорными свойствами, уменьшают силу кислот, и в гомологическом ряду предельных одноосновных карбоновых кислот наиболее сильной является муравьиная кислота. Сила кислот увеличивается, если углеводородный радикал содержит электроноакцепторные заместители, например, галогены.Сама карбоксильная группа оказывает заметное влияние на связанный с ней углеводородный радикал. Так, в случае предельных кислот это выражается в активировании π-положения, а в случае ароматических кислот карбоксильная группа приводит к дезактивации бензольного кольца.Далее необходимо рассмотреть физические свойства кислот и влияние строения карбоксильной группы на их проявление. Так, хорошая растворимость в воде низших кислот обусловлена образованием водородной связи между молекулами кислоты и воды. Необходимо также отметить, что карбоновые кислоты за счет ассоциации молекул существуют в виде циклических димеров. Такая ассоциация возможна за счет прочных водородных связей, и она определяет высокие температуры плавления и кипения кислот.
44 Хим свкорбоновых кислот. Р корбоновых кислот с нуклеафильными реагентами.1. Реакции карбоновых кислот с нуклеофильными реагентами 1.1 Образование солей с металлами:
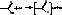
1.2 Взаимодействие с оксидами металлов:

1.3 Взаимодействие с основаниями:

1.4 Взаимодействие с аммиаком:

1.5 Образование галогенангидридов:

1.6 Реакции с Н-нуклеофилами (комплексными гидридами). Комплексные гидриды при взаимодействии с карбоновыми кислотами выделяют водород и образуют карбоксилаты, которые в избытке реагента и при более жестких условиях восстанавливаются до первичных спиртов:

1.7 Реакции с N-нуклеофилами. N-Нуклеофилы (аммиак, амины, гидразин) при взаимодействии с карбоновыми кислотами образуют аммониевые карбоксилаты и при повышенных температурах происходит присоединение к карбонильной группе и получаются амиды:

1.8 Реакции с О-нуклеофилами (реакция этерификации по Фишеру). О-Нуклеофилы нейтрального характера, например, спирты, реагируют и образуют сложные эфиры:

45 Хим св карбоновых кислот. Реакции с электрофильными реагентами. Реакции у α-водородного атома. Галогенирование. Реакции с электрофильными реагентами. Карбоновые кислоты, как очень слабые нуклеофилы, взаимодействуют только с особенно сильными реагентами, присоединение происходит по кислородному атому. Для осуществления реакции с более слабыми электрофильными реагентами необходимо карбоксильную группу активировать, что осуществляется превращением ее в карбоксилат-ион.
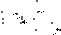
Реакции а-водородного атома
Галогенирование кислот. Подобно альдегидам и кетонам у карбоновых кислот водород у а-углеродного атома приобретает повышенную подвижность. Однако поскольку величина δ+ на атоме углерода карбоксильной группы понижена за счет +М эффекта, то влияние карбоксила на а-углеродный атом значительно меньше, чем у альдегидов и кетонов.Благодаря подвижности атомов водорода связи Са-Н карбоновые кислоты могут подвергаться свободнорадикальному галогенированию. Однако в этом случае вследствие высокой активности и малой избирательности атомов хлора галогенирование может происходить и по другим положениям Цепи
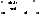
Реакция Геля-Фольгарда-Зелинекого Осуществляют галогеном в присутствии небольших количеств красного фосфора:

6. Реакции декарбоксилирования. Одноосновные кислоты довольно устойчивы к нагреванию. Однако если их нагревать при температуре выше 300 °С с Мп02 или Тh02, то происходит декарбоксилирование с образованием альдегидов или кетонов. Если в углеводородном остатке кислоты имеются сильные элекгроноакцепторные группы, то возможно довольно легкое отщепление С02 при температурах 100-150 °С.
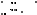
Где X=-NO2,-COOH,-C=0
6.1 Реакция Кольбе
6.5. Дегидратация карбоновых кислот протекает при высоких температурах с образованием кетенов:

6.6 Реакция Дюма

α-аминокислоты, кроме глицина NН2-CH2-COOH.
46.Аминокислоты, изомерия, номенкл. — органические амфотерные соединения, в состав которых входят карбоксильные группы – СООН и аминогруппы -NH2.Аминокислоты можно рассматривать как карбоновые кислоты, в молекулах которых атом водорода в радикале замещен аминогруппой. Классифицируют по структурным признакам. 1. В зависимости от взаимного расположения амино- и карбоксильной групп аминокислоты подразделяют на α-, β-, γ-, δ-, ε- и т. д.
2. В зависимости от количества функциональных групп различают кислые, нейтральные и основные.3. По характеру углеводородного радикала различают алифатические (жирные), ароматические,серосодержащие и гетероциклическиеаминокислоты. Приведенные выше аминокислоты относятся к жирному ряду. НОМЕНКЛАТУРА.По систематической номенклатуре названия аминокислот образуются из названий соответствующих кислот прибавлением приставки амино- и указанием места расположения аминогруппы по отношению к карбоксильной группе. Нумерация углеродной цепи с атома углерода карбоксильной группы.
Например:
Часто используется также другой способ построения названий аминокислот, согласно которому к тривиальному названию карбоновой кислоты добавляется приставка амино- с указанием положения аминогруппы буквой греческого алфавита.
Для α-аминокислот R-CH(NH2)COOH. ИЗОМЕРИЯ1. Изомерия углеродного скелета
2. Изомерия положения функциональных групп
3. Оптическая изомерия --- α-аминокислоты, кроме глицина NН2-CH2-COOH.
47.Синтез аминокислот. Получение, аминирование 1. Действие аммиакана галоидзамещенныежирные кислоты:
реакциюведут с очень большим избыткомаммиакаили в присутствиикарбоната аммония, который защищаетаминогруппу. Образующееся карбаминовое производноеаминокислоты
2.Получение из циангидриновальдегидови кетонов (циангидринный метод). Важной реакциейполучения α-аминокислот является действиеаммиаканациангидриныальдегидови кетонов
с последующим омылениемаминонитрилаваминокислоту:
Аминонитрилыможно получить также, действуя наальдегидыиликетонынепосредственно цианистымаммонием. Этареакцияприобрела особое значение после того, как Н. Д. Зелинский с сотрудниками показали, чтосинильную кислотуиаммиакили цианистыйаммонийможно заменить смесью водныхрастворовхлористогоаммонияицианистого калия, в результате обменного разложения дающих цианистыйаммоний, который и вступает вреакциюсальдегидамииликетонами:
3. Синтезы из сложных эфиров, содержащих подвижныйатомводорода. Большое значение приобрели синтезы аминокислотиз малонового, циануксусного иацетоуксусного эфиров. Пользуясь этими методами, можно получитьаминокислоты, содержащие различные радикалы. а)Синтез из малонового эфира.Из малонового эфираполучают нитрозомалоновый эфир, который восстанавливают в аминомалоновый эфирводородомв присутствиикатализатораилицинкомв кислой среде:
Защитив аминогруппуацетилированием
полученный ацетиламиномалоновый эфир алкилируют, действуя на него последовательно металлическим натриемигалоидным алкилом:
После омыленияидекарбоксилированияполучается α-аминокислота:
б) Синтез из циануксусного эфира проводится аналогично синтезу из малонового эфира.в)Синтез из ацетоуксусного эфира.При действии на однозамещенные ацетоуксусные эфирырастворомфенилдиазотата, а затемщелочьюотщепляетсяуксусная кислотаи образуется неустойчивоеазосоединение, которое изомеризуется в фенилгидразон соответствующейкетокислоты:
Восстановлениефенилгидразонакетокислотыцинкомв солянокислой спиртовой среде приводит к α-аминокислоте:
4. Восстаноление оксимовилигидразоновальдегидо- иликетонокислот.Например:
5. Синтез аминокислотиз фурановых производных (метод А. П. Терентьева и Р. А. Грачевой) основан на легкости окисленияфуранового кольцаперманганатомс образованиемкарбоксильной группы. Если в боковой цепи у фуранового кольца имеетсяаминогруппа(обычно защищенная бензоильной группой), то в результатеокисленияполучается бензоиламинокислота, а послеомыления— самааминокислота. В зависимости от положенияаминогруппыв цепи получается α-, β-, γ- и т. п.кислота.Так, приокислении1-бензоиламино-1-(α-фурил)-алкилов получается α-кислота, например из 1-бензоиламино-1-(α-фурил)-этана —бензоил-α-аланин:
Для получения β-аминокислот пользуются 2-бензоиламино-1-(α-фурил)алкилами
48.Хим. св-ва ам-т. Амфотерность и образование биполярных ионов. Соли с кислотами и основаниями. Аминокислоты-такие производные кислот, к-рые получаются в результате замещения одного или более атомов водорода в радикалах кислот на одну или бол. аминогруппу.Формула ам-ты:
COOH
R〈
NH2
Ам-ты - тв. кристаллические в-ва, хорошо растворимые в воде. Хим. св-ва: вследствие наличия в молекулах ам-т одновременно карбоксильных и аминогрупп ам-ты могут реагировать как кислоты и как амины. Ам-ты – амфотерные соединения. Благодаря наличию карбоксильных групп они способны проявлять кислотные св-ва. В то же время в них имеются аминогруппы, способные присоединять ионы водорода, превращаясь в группы замещённого аммония. Поэтому ам-ты подобно аминам могут проявлять основные св-ва.Образование внутренних солей.Ион водорода, отщепляющийся от карбоксильной группы, присоед-ся к аминогруппе, к-рая превращается как бы в ион замещённого аммония:
H O H O
R - C - C ⪕ ⟶ R - C - C⪕
∣ OH ∣ O
HN ↲ NH3
H
Молекула внутренней соли имеет в двух местах противоположные заряды и как бы включает 2 иона: полож-ый в виде группы NH3+ и отр-ый в виде группы COO-. Поэтому молекулу внутренней соли ам-ты называют биполярным ионом, т.е. ионом, имеющим 2 противоположных заряда.Образование солей.Ам-ты, будучи амфотерными соединениями, образуют соли как с кислотами, так и с осн-ми. В первом случае в реакции участвует аминогруппа, во втором - карбоксильная группа. С основаниями в результате замещения водорода в карбоксиле на металл ам-ты образуют соли. Из них особенно характерны медные соли, кристаллизующиеся обычно в виде крупных кристаллов тёмно-синего цвета. В таких солях ион меди связан дополнительными валентностями с аминогруппами, образуя комплексный ион тёмно-синего цвета. Строение медных солей ам-т выражается формулой:
O
H2N - CH2 - C ⪕
\ O
\ /
Cu
/ \
/ \
O
H2N – CH2 – C ⩽
O
Ам-ты дают соли с к-ми, напр.,
O O
H2N - CH2 - C⪕ +HCl ⟶ Cl [H3N - CH2 - C⪕ ]
OH OH
49. Хим. св-ва аминокислот. Р с одновременным участием карбоксильной и аминогруппы,полипептидаы. Аминокислоты – соединения, в молекулах которых одновременно присутствуют амино- и карбоксильные группы. Общая формула молекул аминокислот - NH2—R—COOH, где R - двухвалентный радикал. Общая формула предельных аминокислот с одной карбоксильной и одной аминогруппой - CnH2n+1NO2. Химические свойства.Реакции по карбоксильным группам А., аминогруппа к-рых защищена ацилированием или солеобразованием, протекают аналогично превращениям карбоновых к-т. А. легко образуют соли, сложные эфиры, амиды, гидразиды, азиды, тиоэфиры, галогенангидриды, смешанные ангидриды и т.д. Эфиры А. под действием натрия или магнийорг. соед. превращаются в аминоспирты. При сухой перегонке в присут. Ва(ОН)2 А. декарбоксилируются. Реакции аминогрупп А. аналогичны превращениям аминов. А. образуют соли с минер, к-тами и пикриновой к-той, легко ацилируются хлорангидридами к-т в водно-щелочном р-ре (р-ция Шоттена - Баумана) и алкилируются алкилгалогенидами. Метилиодид и диазометан превращают А. в бетаины (CH3)3NCHRCOO. С формалином А. дают метилольные или метиленовые производные, а в присут. муравьиной к-ты или каталитически активированного Н2 - N,N-диметиламинокислоты. Под действием HNO2 ароматич. аминогруппы диазотируются, а алифатические замещаются на гидроксил. При обработке эфиров А. изоцианатами и изотиоцианатами образуются производные мочевины и тиомочевины. При нагр. с содой или при одноврем. воздействии алкоголята и СО2 А. дают соли или эфиры N-карбоксипроизводных А., а при использовании CS2 - аналогичные дитиокарбаматы. Реакции с одновременным участием групп NH2 и СООН наиболее характерны для альфа-аминоксилот, к-рые способны образовывать устойчивые 5-членные гетероциклы. С ионами переходных металлов (Си, Zn, Ni, Co, Pb, Ag, Hg, Cr) альфа-А. образуют прочные хелатные комплексы, что используется в комплексонах и в комплексообразующих ионообменных смолах на основе аминокарбоновых и аминофосфоновых к-т. При взаимод. с фосгеном альфа-А. превращаются в циклич. ангидриды N-карбоксиаминокислот, а при нагр. с уксусным ангидридом или ацетилхлоридом - в азлактоны; нагревание А. с мочевиной или обработка изоцианатами дает гидантоины, а при использовании арилизотиоцианатов - тиогидантоины. Вследствие наличия в молекулах ам-т одновременно карбоксильных и аминогрупп ам-ты могут реагировать как кислоты и как амины.Ам-ты – амфотерные соединения. Благодаря наличию карбоксильных групп они способны проявлять кислотные св-ва. В то же время в них имеются аминогруппы, способные присоединять ионы водорода, превращаясь в группы замещённого аммония. Поэтому ам-ты подобно аминам могут проявлять основные св-ва.Образование ангидридов. Подобно оксикислотам, ам-ты легко выделяют воду и дают ангидридоподобные соединения. Из 2-ух молекул α-аминокислот может выделиться либо 1 молекула воды с образованием неполного ангидрида, либо 2 молекулы воды с образованием полного ангидрида. При выделении 1 молекулы воды образуются неполные ангидриды – дипептиды:
H O H O
R – C – C⪕ -H2O R -C - C⪕ OH
∣ OH + ⟶ HN 〈 NH2
HNH C - C〈
HO NH2 // H R
⩾C – C 〈 O
O H R
Дипептиды содержат одну карбоксильную группу и 1 свободную аминогруппу. Поэтому молекула дипептида опять может реагировать с молекулой ам-ты с выделением воды и образованием неполного ангидрида.
O O O O
R // R // H R O -H2O R ∣∣ R ∣∣
H2N – C – C – N – C – C – OH+HN – C - C⪕ ⟶ H2N – C – C – N – C – C -N
H H H H OH H H H H
Дипептид аминокислота трипептид
R O
– C – C ⪕
H OH
Трипептид может реагировать ещё с одной молекулой ам-ты и превращаться в тетрапептид – в-ва, содержащие 4 остатка ам-ты и т.д. В-ва, построенные из многих остатков ам-т – полипептиды. Для всех пептидов характерна пептидная связь, т.е. группа. К группе полипептидов относятся нек. гормоны и широко применяющиеся антибиотики.
O
-C⪕
NH. РЕАКЦИЯ РУЭМАННА (НИНГИДРИНОВАЯ РЕАКЦИЯ (1911)) a-Аминокислоты реагируют с нингидрином, образуя сине-фиолетовый комплекс (пурпур Руэманна), интенсивность окраски которого пропорциональна количеству аминокислоты.Реакция идет по схеме:
Реакция с нингидрином используется для визуального обнаружения a-аминокислот на хроматограммах (на бумаге, в тонком слое), а также для колориметрического определения концентрации аминокислот по интенсивности окраски продукта реакции.Описание опыта. В пробирку наливают 1 мл 1%-го раствора глицина и 0,5 мл 1%-го раствора нингидрина. Содержимое пробирки осторожно нагревают до появления сине-фиолетового окрашивания.
50Углеводы.Классиф. номенклатуры. Изомерии.Состав углеводов В их состав входят углерод (С), водород (Н) и кислород (О). Общая формула углеводов Сn(Н2О)n, где n не меньше трех.Классификация и строение углеводов.Моносахариды (монозы) — простые сахара (альдегиды и кетоны), состоящие из одной молекулы, которые нельзя подвергнуть гидролизу. Они хорошо растворяются в воде и сладкие на вкус. В зависимости от числа атомов углерода, входящих в состав молекул моносахаридов. Различают следующие моносахариды, широко представленные в клетках: 1) триозы (молочная кислота, пировиноградная кислота — Сз); 2) тетрозы (эритроза — С4);3) пентозы (рибоза, дезоксирибоза — С5); 4) гексозы (глюкоза, фруктоза, галактоза — С6). Дисахариды — два моносахарида, соединенных вместе гликозидной связью. Они хорошо растворимы в воде и сладкие на вкус. Наиболее широкое распространение имеют: 1) сахароза (глюкоза+фруктоза); 2) лактоза (глюкоза+галактоза); 3) мальтоза (глюкоза+глюкоза). Полисахариды (полиозы) — полимеры регулярного строения, состоящие из моносахаридов. Они не растворимы в воде, поэтому не сладкие на вкус. Способны гидролизоваться до моносахаридов. Различают: 1) гомополисахариды, имеющие в составе моносахариды одного вида (крахмал, гликоген, целлюлоза состоят из молекул глюкозы (С6Н10О5)n); 2) гетерополисахариды, имеющие в составе моносахариды различных видов и их производные (гемицеллюлоза состоит из различных гексоз и пентоз, гликопротеиды и гликолипиды — из углеводов, белков и липидов).Оптическая изомерияЕсли атом углерода в молекуле связан с четырьмя различными атомами или атомными группами, например:
то возможно существование двух соединений с одинаковой структурной формулой, но отличающихся пространственным строением. Молекулы таких соединений относятся друг к другу как предмет и его зеркальное изображение и являются пространственными изомерами.Изомерия этого вида называется оптической, изомеры – оптическими изомерами или оптическими антиподами:
Молекулы оптических изомеров несовместимы в пространстве (как левая и правая руки), в них отсутствует плоскость симметрии. Таким образом, оптическими изомерами называются пространственные изомеры, молекулы которых относятся между собой как предмет и несовместимое с ним зеркальное изображение.Оптические изомеры имеют одинаковые физические и химические свойства, но различаются отношением к поляризованному свету. Такие изомеры обладают оптической активностью (один из них вращает плоскость поляризованного света влево, а другой - на такой же угол вправо). Различия в химических свойствах наблюдаются только в реакциях с оптически активными реагентами.Оптическая изомерия проявляется в органических веществах различных классов и играет очень важную роль в химии природных соединений.
51.Углеводы. Антиподы, диастеомеры..D-ряд, L-ряд, связь с альдегидом. Состав углеводов В их состав входят углерод (С), водород (Н) и кислород (О). Общая формула углеводов Сn(Н2О)n, где n не меньше трех.Классификация и строение углеводов.Моносахариды (монозы) — простые сахара (альдегиды и кетоны), состоящие из одной молекулы, которые нельзя подвергнуть гидролизу. Они хорошо растворяются в воде и сладкие на вкус. В зависимости от числа атомов углерода, входящих в состав молекул моносахаридов. Различают следующие моносахариды, широко представленные в клетках: 1) триозы (молочная кислота, пировиноградная кислота — Сз); 2) тетрозы (эритроза — С4);3) пентозы (рибоза, дезоксирибоза — С5); 4) гексозы (глюкоза, фруктоза, галактоза — С6). Дисахариды — два моносахарида, соединенных вместе гликозидной связью. Они хорошо растворимы в воде и сладкие на вкус. Наиболее широкое распространение имеют: 1) сахароза (глюкоза+фруктоза); 2) лактоза (глюкоза+галактоза); 3) мальтоза (глюкоза+глюкоза). Полисахариды (полиозы) — полимеры регулярного строения, состоящие из моносахаридов. Они не растворимы в воде, поэтому не сладкие на вкус. Способны гидролизоваться до моносахаридов. Различают: 1) гомополисахариды, имеющие в составе моносахариды одного вида (крахмал, гликоген, целлюлоза состоят из молекул глюкозы (С6Н10О5)n); 2) гетерополисахариды, имеющие в составе моносахариды различных видов и их производные (гемицеллюлоза состоит из различных гексоз и пентоз, гликопротеиды и гликолипиды — из углеводов, белков и липидов).Диастереомерия- стеомеря, обусловленная различным расположением заместителей в пространстве относительно плоскости. Условия нобходимые:-наличие пи-связи- у атомов углнрода, соеденнывх двойной связью наличичие разных заместителей.Диастериомеры – пара стереомеров, у которых конфигурация 1 хиральных атомов одинаковая, у других противоположная.Антиподы- пара стериомеров, у которых конфигурация у всех хиральных ценров с один.ю окружением противолположная.Тауторемия- вид структурной изомерии, при которой изомеры превращаются друг в друга путем перемещения атомов или групп внутри молекул. Превращение требует спрециальных условий.Кольчато*цепная:
D-ряд – ряд стериоизомеров, имеющих конфигурацию, сходную с конфигурацией D—глицеринового альдегида.(водород слева)L-ряд- ряд стериоизомеров, имеющих конфигурацию, сходную с конфигурацией L—глицеринового альдегида.(водород справа)
Глюкоза
D-ряд - ряд стериоизомеров, имеющих конфигурацию, сходную с конфигурацией D-глицеринового альдегида.(водород слева) L-ряд- ряд стериоизомеров, имеющих конфигурацию, сходную с конфигурацией L-глицеринового альдегида.(водород справа)
52.Углеводы. Изомерия, связанная с наличием пиранозного и фуранозного цикла. Конформационная изомерия. Состав углеводов В их состав входят углерод (С), водород (Н) и кислород (О). Общая формула углеводов Сn(Н2О)n, где n не меньше трех.Классификация и строение углеводов.Моносахариды (монозы) — простые сахара (альдегиды и кетоны), состоящие из одной молекулы, которые нельзя подвергнуть гидролизу. Они хорошо растворяются в воде и сладкие на вкус. В зависимости от числа атомов углерода, входящих в состав молекул моносахаридов. Различают следующие моносахариды, широко представленные в клетках: 1) триозы (молочная кислота, пировиноградная кислота — Сз); 2) тетрозы (эритроза — С4);3) пентозы (рибоза, дезоксирибоза — С5); 4) гексозы (глюкоза, фруктоза, галактоза — С6). Дисахариды — два моносахарида, соединенных вместе гликозидной связью. Они хорошо растворимы в воде и сладкие на вкус. Наиболее широкое распространение имеют: 1) сахароза (глюкоза+фруктоза); 2) лактоза (глюкоза+галактоза); 3) мальтоза (глюкоза+глюкоза). Полисахариды (полиозы) — полимеры регулярного строения, состоящие из моносахаридов. Они не растворимы в воде, поэтому не сладкие на вкус. Способны гидролизоваться до моносахаридов. Различают: 1) гомополисахариды, имеющие в составе моносахариды одного вида (крахмал, гликоген, целлюлоза состоят из молекул глюкозы (С6Н10О5)n); 2) гетерополисахариды, имеющие в составе моносахариды различных видов и их производные (гемицеллюлоза состоит из различных гексоз и пентоз, гликопротеиды и гликолипиды — из углеводов, белков и липидов).Оптическая изомерияЕсли атом углерода в молекуле связан с четырьмя различными атомами или атомными группами, например:
то возможно существование двух соединений с одинаковой структурной формулой, но отличающихся пространственным строением. Молекулы таких соединений относятся друг к другу как предмет и его зеркальное изображение и являются пространственными изомерами.Конформационная изомерия обусловлена возможностью свободного вращения вокруг одинарных связей, без существенного изменения геометрии и большого числа поворотных или комформационных изомеров.Виды:1.торисионное напряжение 2. Напряжение Ван-дер-Вальса, 3. Угловое напряжение Конформер молекула в конормации в которую ее атомы самопроизвольно возращаются после небольших сдвигов. С удлинением углеродных цепей возникают различные конформации цепи.Конформация циклов возникает за счет изменения колебательных состояний атомов и атомных группировок, что создает возможность образования многих неустойчивых форм.