

2009.-Byelorussian Pharmacopoeia_Volume 3
.pdfОбщие статьи на лекарственные формы и субстанции |
121 |
ОБЩИЕ СТАТЬИ НА ЛЕКАРСТВЕННЫЕ ФОРМЫ И СУБСТАНЦИИ
ЛЕКАРСТВЕННЫЕ СРЕДСТВА НА ОСНОВЕ ЛЕКАРСТВЕННОГО РАСТИТЕЛЬНОГО СЫРЬЯ
# ЛЕКАРСТВЕННОЕ РАСТИТЕЛЬНОЕ СЫРЬЕ
ОПРЕДЕЛЕНИЕ
Лекарственное растительное сырье — это
цельные растения, фрагменты растений или измельченные растения, части растений, мор-
ские водоросли, грибы, лишайники в непереработанной форме, обычно высушенные, а иногда и в свежем виде. Некоторые эксудаты, которые не были подвергнуты специальной обработке,
также могут относиться к лекарственному расти-
тельному сырью. Лекарственное растительное
сырье точно определяется его ботаническим на-
учным названием и в соответствии с имеющейся
биноминальной системой (род, вид, разновид-
ность и автор).
ПРОИЗВОДСТВО
Лекарственное растительное сырье полу-
чают из специально выращиваемых или дико-
растущих растений. Чтобы гарантировать вы-
сокое качество лекарственного растительного сырья, важно соблюдать соответствующие правила выращивания, сбора урожая, сушки, измельчения и условий хранения.
Лекарственное растительное сырье должно быть по возможности тщательно очищено от примесей, таких как остатки почвы, пыли, грязи, загрязнений наподобие грибов,
насекомых и других примесей животного про-
исхождения. Оно не должно быть подгнившим.
Если проводилась деконтаминация, необходимо убедиться, что части растения не пострадали и что после обработки в сырье не остались вредные примеси. Для деконтаминации лекарственного растительного сырья запрещается использование этиленоксида.
ИДЕНТИФИКАЦИЯ
Лекарственное растительное сырье идентифицируют с использованием его макроскопических и микроскопических признаков; при необходимости могут применяться и некоторые другие методы испытаний (например, тонкослойная
хроматография).
ИСПЫТАНИЯ
Допустимые примеси (#2.8.2). Если иного
не указано в частной статье, проводят испытание на допустимые примеси. Количество допустимых примесей указывают в частной статье.
Потеря в массе при высушивании (2.2.32). Если иного не указано в частной статье, проводят испытание на потерю в массе при высушивании.
Вода (2.2.13). Определение воды может проводиться вместо испытания «Потеря в массе
при высушивании» для лекарственного расти-
тельного сырья с высоким содержанием эфирных масел.
Общая зола (2.4.16).
Пестициды (2.8.13). Лекарственное растительное сырье должно соответствовать требованиям в отношении остаточных количеств пестицидов. Эти требования применяются с учетом происхождения растения, его дальнейшего ис-
пользования с учетом протокола полных данных
по обработке определенной партии растения. Содержание остатков пестицидов может быть определено посредством метода, описанного в приложении к основному методу.
Микробиологическая чистота. Рекомендации по исследованию микробиологической чистоты продуктов, состоящих из одного или нескольких видов лекарственного растительного сырья, приводятся в разделе 5.1.4. Микробиологическая чистота лекарственных средств (Категория 4).
Тяжелые металлы. Необходимо учитывать также риск загрязнения лекарственного расти-
тельного сырья тяжелыми металлами. Если в частной статье не указывается предельное содержание тяжелых металлов или других отдель-
ных элементов, такие предельные нормы при необходимости могут быть установлены. Если иного не указано в частной статье, лекарственное растительное сырье должно соответствовать нормам и требованиям действующего законодательства по содержанию тяжелых металлов.
Радиоактивное загрязнение. Если иного не указано в частной статье, лекарственное растительное сырье должно соответствовать
нормам и требованиям действующего законодательства по содержанию радионуклидов.
При необходимости лекарственное расти-
тельное сырье подвергают, например, нижеследующим испытаниям.
Зола, нерастворимая в хлористоводородной кислоте (2.8.1).
Экстрактивные вещества.
Коэффициент набухания (2.8.4).
Показатель горечи (2.8.15).
Афлатоксины.
122 |
Государственная фармакопея Республики Беларусь |
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Если нет специальных требований, количественное содержание устанавливают с использованием подходящего метода.
ХРАНЕНИЕ
В защищенном от влаги и света месте при температуре от 15°С до 25°С, если иного не указано в частной статье.
Частные фармакопейные статьи на субстанции |
123 |
ЧАСТНЫЕ ФАРМАКОПЕЙНЫЕ СТАТЬИ НА СУБСТАНЦИИ ДЛЯ ФАРМАЦЕВТИЧЕСКОГО ИСПОЛЬЗОВАНИЯ
ВВЕДЕНИЕ
Субстанции для фармацевтического ис-
пользования подразделяются на фармацев-
тические субстанции и вспомогательные вещества. Качество субстанций для фармацевтического использования регламентируется
требованиями соответствующей частной фар-
макопейной статьи Государственной фарма-
копеи Республики Беларусь и/или норматив-
ной документацией (фармакопейной статьей ФС) для отечественных производителей или
нормативным документом (НД) для зарубеж-
ных производителей), утвержденной уполномоченным органом.
Если субстанция данного производителя имеет сертификат соответствия частной статье
Европейской Фармакопеи или аналогичное
разрешение уполномоченного органа, ее качество может контролироваться непосредственно соответствующей частной статьей ГФ РБ, а в случае его отсутствия качество субстанций контролируется по НД, утвержденной уполномоченным органом. При этом требования НД к качеству субстанции должны быть не ниже требований соответствующей частной фармакопейной статьи ГФ РБ.
Требования частной фармакопейной статьи ГФ РБ не всегда могут быть достаточным условием для окончательного заключения о качестве субстанции конкретного производи-
теля — в таком случае необходимо принимать во внимание производство субстанции в соответствии с требованиями Надлежащей производственной практики по известной технологии, условия реализации. Все изменения, вно-
симые в технологию производства субстанций,
освоение старой или разработка новой техно-
логии производства другими производителя-
ми должны сопровождаться представлением в
уполномоченный орган данных, подтверждаю-
щих возможность контроля качества этой субстанции по соответствующей частной статье
ГФ РБ или с внесением в нее дополнительных
требований по необходимым разделам либо по
вновь разработанному и утвержденному НД. Субстанция может использоваться в
период всего срока годности, если ее каче-
ство соответствует утвержденной спецификации, но может также использоваться после окончания срока годности при условии ее переконтроля.
Дата переконтроля — это дата, после ко-
торой образцы субстанции должны быть иссле-
дованы на предмет соответствия специфика-
ции и возможности использования в производ-
стве данного лекарственного средства.
Период переконтроля — период, в течение которого субстанция для фармацевтиче-
ского использования считается соответствующей спецификации и, следовательно, пригод-
ной для производства данного лекарственного средства, если она хранилась в рекомендованных условиях. По истечении данного пери-
ода серия должна быть повторно испытана на
соответствие спецификации и затем незамед-
лительно использована.
Переконтроль допускается для синтетических субстанций, характеризующихся зна-
чительной стабильностью. Для субстанций
биологического происхождения устанавливается только срок годности, по истечении ко-
торого субстанции использованию не подле-
жат. Переконтроль допускается для некото-
рых определенных антибиотиков.
АДЕНОЗИН
Adenosinum
ADENOSINE
NH2
N
N
HO N N
O
OH OH
C10H13N5О4 |
М.м. 267,2 |
ОПРЕДЕЛЕНИЕ
Аденозин содержит не менее 99,0 % и не более 101,0 % 9-β-D-рибофуранозил-9Н- пурин-6-амина в пересчете на сухое веще-
ство.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Малорастворим в воде, растворим в горячей
воде, практически нерастворим в 96% спирте и
в метиленхлориде. Растворяется в разведенных
минеральных кислотах.
Температура плавления: около 234°С.
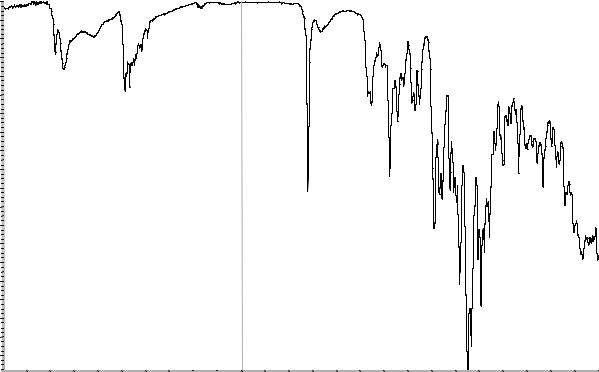
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
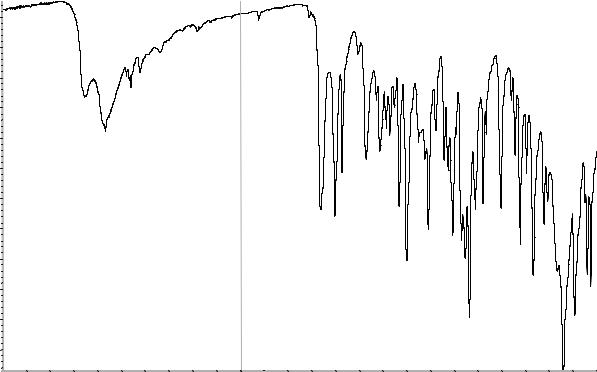
Абсорбционная спектрофотометрия в ин-
фракрасной области (2.2.24).
Сравнение: ФСО аденозина # или спектр,
представленный на рисунке 1.
124 |
Государственная фармакопея Республики Беларусь |
Пропускание
95
90
85
80
75
70
65
60
55
50
45
40
3000 |
2000 |
1500 |
1000 |
|
Волновое |
число (см-1) |
|
Рисунок 1. Инфракрасный спектр пропускания ФСО аденозина.
ИСПЫТАНИЯ |
Раствор сравнения (а). 1,0 мл испытуемого |
Раствор S. 5,0 г испытуемого образца су- |
раствора доводят подвижной фазой до объема |
спендируют в 100 мл воды дистиллированной Р |
100,0 мл. 1,0 мл полученного раствора доводят |
и нагревают до кипения. Охлаждают, фильтруют |
подвижной фазой до объема 10,0 мл. |
под вакуумом и доводят водой дистиллирован- |
Раствор сравнения (b). 5 мг аденина Р |
ной Р до объема 100 мл. |
(примесь А) и 5 мг инозина Р (примесь G) раст- |
Цветность (2.2.2, метод II). Раствор S |
воряют в подвижной фазе и доводят до объема |
должен быть бесцветным. |
50 мл этим же растворителем. 4 мл получен- |
Кислотность и щелочность. К 10 мл раст- |
ного раствора доводят подвижной фазой до |
вора S прибавляют 0,1 мл раствора бромкре- |
объема 100 мл. |
золового пурпурового Р и 0,1 мл 0,01 М раст- |
Условия хроматографирования: |
вора кислоты хлористоводородной. Появляет- |
– колонка длиной 0,25 м и внутренним диа- |
ся желтое окрашивание. При прибавлении не |
метром 4,6 мм, заполненная силикагелем окта- |
более 0,4 мл 0,01 М раствора натрия гидрок- |
децилсилильным эндкепированным для хрома- |
сида должно появиться фиолетово-синее окра- |
тографии Р с размером частиц 5 мкм; |
шивание. |
– подвижная фаза: вода Р — смесь раство- |
Удельное оптическое вращение (2.2.7). От |
рителей (40:60, об/об); |
-45 до -49 в пересчете на сухое вещество. 1,25 г |
– скорость подвижной фазы: 1,5 мл/мин; |
испытуемого образца растворяют в 1 М раство- |
– спектрофотометрический детектор, |
ре кислоты хлористоводородной и доводят до |
длина волны 254 нм; |
объема 50,0 мл этим же растворителем. Полу- |
– объем вводимой пробы: 20 мкл; |
ченный раствор используют в течение 10 мин |
– время хроматографирования: 1,5-крат- |
после приготовления. |
ное время удерживания аденозина. |
Сопутствующие примеси. Жидкостная |
Относительное удерживание (по отноше- |
хроматография (2.2.29). |
нию к аденозину; время удерживания — около |
Смесь растворителей. 6,8 г калия гидро- |
13 мин): примесь А — около 0,3; примесь G — |
сульфата Р и 3,4 г тетрабутиламмония ги- |
около 0,4. |
дросульфата Р растворяют в воде Р, доводят |
Пригодность хроматографической систе- |
до рН 6,5 раствором 60 г/л калия гидроксида Р и |
мы: раствор сравнения (b): |
разводят водой Р до объема 1000 мл. Использу- |
– разрешение: не менее 1,5 между пиками |
ют свежеприготовленную смесь растворителей. |
примеси А и примеси G. |
Испытуемый раствор. 20 мг испытуемого |
Предельное содержание примесей (для рас- |
образца растворяют в подвижной фазе и дово- |
чета содержания примесей умножают площади |
дят до объема 20 мл этим же растворителем. |
пиков на соответствующие поправочные коэф- |
Адреналина тартрат (# эпинефрина гидротартрат) |
125 |
фициенты: для примеси А — 0,6; для примеси
G — 1,4):
–примесь А (не более 0,2 %): на хромато-
грамме испытуемого раствора площадь пика, соответствующего примеси А, не должна превышать 2-кратную площадь основного пика на хроматограмме раствора сравнения (а);
–примесь G (не более 0,1 %): на хроматограмме испытуемого раствора площадь пика, соответствующего примеси G, не должна пре-
вышать площадь основного пика на хроматограмме раствора сравнения (а);
–неспецифицированные примеси (не более 0,10 %): на хроматограмме испытуе-
мого раствора площадь любого пика, кроме
основного и пиков примесей А и G, не должна превышать площадь основного пика на хро-
матограмме раствора сравнения (а);
–сумма примесей (не более 0,5 %): на
хроматограмме испытуемого раствора сумма площадей всех пиков, кроме основного, не
должна превышать 5-кратную площадь основного пика на хроматограмме раствора сравне-
ния (а).
На хроматограмме испытуемого раствора не учитывают пики с площадью менее 0,5 пло-
щади основного пика на хроматограмме раст-
вора сравнения (а) (0,05 %).
Хлориды (2.4.4). Не более 0,01 % (100 ppm). 10 мл раствора S доводят водой Р до объема 15 мл. Полученный раствор
должен выдерживать испытание на хлориды.
Сульфаты (2.4.13). Не более 0,02 % (200 ppm). 15 мл раствора S должны выдерживать испытание на сульфаты.
Аммония соли (2.4.1, метод В). Не более 0,001 % (10 ppm). 0,5 г испытуемого образца должны выдерживать испытание на соли аммония. Эталон готовят с использова-
нием 5 мл эталонного раствора аммония (1 ррm NH4) Р.
Потеря в массе при высушивании
(2.2.32). Не более 0,5 %. 1,000 г испытуемого
образца сушат при температуре 105°С.
Сульфатная зола (2.4.14, метод А). Не более 0,1 %. Определение проводят из 1,0 г
испытуемого образца.
#Остаточные количества органических растворителей (2.4.24). Испытуемый обра-
зец должен выдерживать требования статьи (5.4).
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Аденозин в условиях испытания
не обладает антимикробным действием.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,200 г испытуемого образца растворяют, при необходимости слегка подогревая, в смеси из 20 мл уксусного ангидрида Р и 30 мл кислоты уксусной безводной Р и титруют 0,1 М раствором хлорной кислоты потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соот-
ветствует 26,72 мг C10H13N5O4.
ПРИМЕСИ
Специфицированные примеси: A, G. Другие обнаруживаемые примеси (следу-
ющие вещества, если они присутствуют в зна-
чительных количествах, следует определять
тем или иным испытанием, описанным в частной статье. Их содержание лимитируется общим
критерием приемлемости для других/неспе-
цифицированных примесей и/или общей ста-
тьей Субстанции для фармацевтического использования. Вследствие этого нет необходимости идентифицировать эти примеси для доказа-
тельства соответствия требованиям. См. также
статью 5.10. Контроль примесей в субстанциях для фармацевтического использования): F, H.
А. Аденин.
O
HN
HO O N
O
OH OH
F. 1-β-D-Рибофуранозилпиримидин-2,4(1Н,3Н)-
дион (уридин).
O
N
HN
R N N
HO
O
OH OH
G. R = Н: 9-β-D-Рибофуранозил-1,9-дигидро-
6Н-пурин-6-он (инозин).
Н. R = NH2: 2-Амино-9-β-D-рибофуранозил- 1,9-дигидро-6Н-пурин-6-он (гуанозин).
АДРЕНАЛИНА ТАРТРАТ (# ЭПИНЕФРИНА ГИДРОТАРТРАТ)
Adrenalini tartras
ADRENALINE TARTRATE
HOH
HO |
H |
H OH |
|
||
N |
|
CO2H |
|||
|
|
|
CH |
|
|
|
|
|
3 |
HO2C |
|
|
|
|
|
|
|
HO |
|
H |
OH |
||
|
|
|
|||
C9H13NO3 · C4H6O6 |
|
М.м. 333,3 |
|||
ОПРЕДЕЛЕНИЕ |
|
|
|
||
Адреналина |
тартрат |
содержит |
не |
||
менее 98,5% и не более 101,0% (1R)-1-(3,4- дигидроксифенил)-2-(метиламино)этанола гидро(2R,3R)-2,3-дигидроксибутандиоата в пе-
ресчете на сухое вещество.
126 |
Государственная фармакопея Республики Беларусь |
ОПИСАНИЕ (СВОЙСТВА)
Белый или серовато-белый кристаллический порошок.
Легкорастворим в воде, малорастворим в спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Удельное оптическое вращение (2.2.7): от -50 до -53,5. 5,0 г испытуемого образца
растворяют в 50 мл раствора 5 г/л натрия метабисульфита Р и подщелачивают раствором аммиака Р. Полученную смесь выдержи-
вают при комнатной температуре не менее 15 мин и фильтруют. Фильтрат используют для идентификации С. Полученный осадок (адре-
налин) трижды промывают метанолом Р пор-
циями по 10 мл и сушат при температуре 80°С.
Готовят раствор 20,0 г/л полученного адреналина в 0,5 М растворе хлористоводородной кислоты.
В. Абсорбционная спектрофотометрия в
инфракрасной области (2.2.24). Приготовление: в дисках; используют
адреналин, полученный в идентификации А. Сравнение: адреналин, полученный как
указано в идентификации А из 50 мг ФСО адреналина тартрата, растворенного в 5 мл раствора 5 г/л натрия метабисульфита Р. Полученную смесь выдерживают при комнатной температуре не менее 30 мин. Фильтруют через стеклянный фильтр.
С. 0,2 мл фильтрата, полученного в идентификации А, дают реакцию (b) на тартраты (2.3.1).
ИСПЫТАНИЯ
Раствор S. 0,5 г испытуемого образца растворяют в воде Р и доводят до объема 10 мл. Раствор используют немедленно.
Прозрачность (2.2.1). Раствор S по степени
мутности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора S должна быть не интенсивнее эталона BY(КЖ)5.
Сопутствующие примеси. Жидкостная хроматография (2.2.29). Растворы готовят с защитой от света.
Смесь растворителей А. 5,0 г калия дигидрофосфата Р растворяют в воде для хроматографии Р, в полученном растворе растворяют 2,6 г натрия октансульфоната Р и доводят водой для хроматографии Р до объема
1000 мл (для полного растворения компонен-
тов раствор обычно перемешивают в течение
не менее 30 мин). Доводят до рН 2,8 кислотой фосфорной Р.
Смесь растворителей В. Ацетонитрил Р1 — смесь растворителей А (130:870, об/об).
Испытуемый раствор. 75 мг испытуемого
образца растворяют в 5 мл 0,1 М раствора кислоты хлористоводородной и доводят смесью
растворителей В до объема 50 мл.
Раствор сравнения (а). 1,0 мл испытуемо-
го раствора доводят смесью растворителей В до
объема 100,0 мл. 1,0 мл полученного раствора доводят смесью растворителей В до объема 10,0 мл.
Раствор сравнения (b). 1,5 мг ФСО норадреналина тартрата (примесь В) и 1,5 мг ФСО адреналона гидрохлорида (примесь С)
растворяют в смеси растворителей В, прибавляют 1,0 мл испытуемого раствора и доводят смесью растворителей В до объема 100,0 мл.
Раствор сравнения (с). Содержимое контейнера ФСО адреналина смеси примесей (при-
меси D и Е) растворяют в 0,1 мл 0,1 М раствора кислоты хлористоводородной и прибавляют 0,9 мл смеси растворителей В.
Раствор сравнения (d). 7,5 мг ФСО адреналина тартрата с примесью А растворяют в 0,5 мл 0,1 М кислоты хлористоводородной
и доводят смесью растворителей В до объема
5,0 мл.
Холостой раствор. 0,1 М раствор кислоты хлористоводородной — смесь растворителей В (1:9, об/об).
Условия хроматографирования:
–колонка длиной 0,10 м и внутренним диаметром 4,6 мм, заполненная силикагелем октадецилсилильным эндкепированным для хроматографии Р с размером частиц 3 мкм;
–температура: 50°С;
–подвижная фаза:
–подвижная фаза А: ацетонитрил Р1 —
смесь растворителей А (5:95, об/об);
–подвижная фаза В: ацетонитрил Р1 —
смесь растворителей А (45:55, об/об);
Время (мин) |
Подвижная |
Подвижная |
фаза А |
фаза В |
|
|
(%, об/об) |
(%, об/об) |
0—15 |
92 → 50 |
8 → 50 |
15—20 |
50 → 92 |
50 → 8 |
20—25 |
92 |
8 |
|
|
|
–скорость подвижной фазы: 2,0 мл/мин;
–спектрофотометрический детектор, длина волны 280 нм;
–объем вводимой пробы: 20 мкл. Идентификация пиков примесей: иденти-
фицируют пики примесей D и E, используя хроматограмму раствора сравнения (с) и хромато-
грамму, прилагаемую к ФСО адреналина смеси примесей; идентифицируют пик примеси А, ис-
пользуя хроматограмму раствора сравнения (d)
и хроматограмму, прилагаемую к ФСО адреналина тартрата с примесью А.
Относительное удерживание (по отношению к адреналину; время удерживания — около 4 мин): примесь В — около 0,8; примесь С — около 1,3; примесь А — около 3,2; примесь D — около 3,3; примесь Е — около 3,7.
Пригодность хроматографической системы: раствор сравнения (b):
Азитромицин |
127 |
–разрешение: не менее 3,0 между пиками
примеси В и адреналина.
Предельное содержание примесей (для
расчета содержания примесей умножают площади пиков на соответствующие поправочные коэффициенты: для примеси D — 0,7; для примеси Е — 0,6):
–примесь А (не более 0,3 %): на хроматограмме испытуемого раствора площадь пика, соответствующего примеси А, не должна пре-
вышать 3-кратную площадь основного пика на хроматограмме раствора сравнения (а);
–примеси В, С (не более 0,2 %): на хроматограмме испытуемого раствора площади
пиков, соответствующих примесям В и С, не
должны превышать 2-кратную площадь основного пика на хроматограмме раствора сравне-
ния (а);
–примеси D, Е (не более 0,1 %): на хро-
матограмме испытуемого раствора площади пиков, соответствующих примесям D и Е, не
должны превышать площадь основного пика на хроматограмме раствора сравнения (а);
–неспецифицированные примеси (не более 0,10 %): на хроматограмме испытуемого раствора площадь любого пика, кроме
основного и пиков примесей А, В, С, D и Е, не должна превышать площадь основного пика на хроматограмме раствора сравнения (а);
–сумма примесей (не более 0,6 %): на хроматограмме испытуемого раствора сумма площадей всех пиков, кроме основного, не должна превышать 6-кратную площадь основного пика на хроматограмме раствора сравне-
ния (а).
На хроматограмме испытуемого раствора не учитывают пики с площадью менее 0,5 площади основного пика на хроматограмме раствора сравнения (а) (0,05 %).
Потеря в массе при высушивании
(2.2.32). Не более 0,5 %. 1,000 г испытуемого образца сушат в вакууме в течение 18 ч.
Сульфатная зола (2.4.14, метод А). Не
более 0,1 %. Определение проводят из 1,000 г испытуемого образца.
#Остаточные количества органических растворителей (2.4.24). Испытуемый образец
должен выдерживать требования статьи (5.4).
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Адреналина тартрат в условиях испытания обладает антимикробным действи-
ем. Посев на питательные среды проводят ме-
тодом мембранной фильтрации.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,300 г испытуемого образца растворяют
в 50 мл кислоты уксусной безводной Р, при необходимости слегка подогревая, и титруют
0,1 М раствором кислоты хлорной до появления синевато-зеленого окрашивания, используя в качестве индикатора 0,1 мл раствора кристаллического фиолетового Р.
ХРАНЕНИЕ
Ввоздухонепроницаемом контейнере или
взапаянной ампуле под вакуумом или в среде инертного газа в защищенном от света месте.
ПРИМЕСИ
Специфицированные примеси: А, В, С, D, Е.
А. Структура неизвестна.
H OH
HO |
NH2 |
HO
В. Норадреналин.
O R
HO |
N |
CH3
HO
С.R=H:1-(3,4-Дигидроксифенил)-2-(метиламино)-
этанон (адреналон).
Е. R = CH2-C6H5: 2-(Бензилметиламино)-1- (3,4-дигидроксифенил)этанон.
HOH
HO |
N |
CH3
HO
D. (1R)-2-(Бензилметиламино)-1-(3,4-дигидрокси-
фенил)этанол.
АЗИТРОМИЦИН
Azithromycinum
AZITHROMYCIN
|
|
|
|
CH3 |
|
|
|
|
H3C |
N |
CH3 |
|
|
|
H |
H |
H |
|
|
|
H3C |
OH |
|
|
CH3 |
|
HO |
HO |
CH3 |
|
|
|
|
|
|
H3C |
CH3O O |
|
|
CH3 O |
H |
|
N |
|
O H |
H |
CH3 |
|
OH |
|
|||
|
|
O |
|
O |
|
|
OH |
CH3 |
H |
|
|
|
|
|
|||
|
OCH3 |
H CH3 |
|
||
|
|
|
|
||
|
|
|
|
|
x H2O |
CH3
C38H72N2O12·хH2O М.м. 749 (безводное вещество) x = 1 или 2
ОПРЕДЕЛЕНИЕ
Азитромицин содержит не менее 96,0%
и не более 102,0% (2R,3S,4R,5R,8R,10R,11R, 12S,13S,14R)-13-[(2,6-дидезокси-3-С-метил- 3-О-метил-α-L-рибо-гексопиранозил)окси]-2- этил-3,4,10-тригидрокси-3,5,6,8,10,12,14-гепта- метил-11-[[3,4,6-тридезокси-3-(диметиламино)- β-D-ксило-гексопиранозил]окси]-1-окса-6-аза-
циклопентадекан-15-она в пересчете на без-
128 |
Государственная фармакопея Республики Беларусь |
водное вещество. Содержит 1 или 2 молекулы
воды.
Полусинтетический продукт, полученный из
продукта ферментации.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, легкорастворим в этаноле и в метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфракрасной области (2.2.24).
Сравнение: ФСО азитромицина # или
спектр, представленный на рисунке 1.
Если спектры отличаются, то готовят рас-
творы 90 г/л испытуемого образца и ФСО азитромицина в метиленхлориде Р, которые используют для получения новых спектров.
ИСПЫТАНИЯ
Раствор S. 0,500 г испытуемого образца растворяют в этаноле Р и доводят до объема
50,0 мл этим же растворителем. Прозрачность (2.2.1). Раствор S должен
быть прозрачным.
Цветность (2.2.2, метод II). Раствор S должен быть бесцветным.
рН (2.2.3). От 9,0 до 11,0. 0,100 г испытуемого образца растворяют в 25,0 мл метанола Р и
доводят водой, свободной от углерода диоксида, Р до объема 50,0 мл.
Удельное оптическое вращение (2.2.7). От -45 до -49 в пересчете на безводное вещество.
Определение проводят с использованием раст-
вора S.
Сопутствующие примеси. Жидкостная
хроматография (2.2.29).
Смесь растворителей. Раствор 1,73 г/л аммония дигидрофосфата Р доводят до
рН 10,0 раствором аммиака Р. К 350 мл полу-
ченного раствора прибавляют 300 мл ацетонитрила Р1, 350 мл метанола Р1 и тщательно перемешивают.
Испытуемый раствор. 0,200 г испытуемо-
го образца растворяют в смеси растворителей и доводят до объема 25,0 мл этим же растворителем.
Раствор сравнения (a). 1,0 мл испытуемо-
го раствора доводят смесью растворителей до
объема 100,0 мл.
Раствор сравнения (b). Содержимое контейнера с ФСО азитромицина для проверки пригодности хроматографической системы
(содержит примеси F, H и J) растворяют в 1,0 мл
смеси растворителей и обрабатывают ультразвуком в течение 5 мин.
Раствор сравнения (с). 8,0 мг ФСО азитромицина для идентификации пиков (содержит примеси A, B, C, E, F, G, I, J, L, M, N, O, P) раст-
воряют в 1,0 мл смеси растворителей.
Раствор сравнения (d). Содержимое контейнера с ФСО азитромицина примеси В растворяют в 1,0 мл смеси растворителей.
Условия хроматографирования:
– колонкадлиной0,25мивнутреннимдиаме-
тром 4,6 мм, заполненная кремнийорганическим полимером аморфным октадецилсилильным
Пропускание
100
98
96
94
92
90
88
86
84
82
80
78
76
74
72
70
68
66
64
62
3000 |
2000 |
1500 |
1000 |
|
Волновое |
число (см-1) |
|
Рисунок 1. Инфракрасный спектр пропускания ФСО азитромицина.
Азитромицин |
129 |
эндкепированным для масс-спектрометрии Р с
размером частиц 5 мкм;
–температура: 60°С;
–подвижная фаза:
–подвижная фаза А: раствор 1,80 г/л динатрия гидрофосфата безводного Р, доведенный до рН 8,9 кислотой фосфорной разведенной Р или раствором натрия гидроксида разведенным Р;
–подвижная фаза В: метанол Р1 — ацетонитрил Р1 (250:750, об/об);
Время (мин) |
Подвижная |
Подвижная |
фаза А |
фаза В |
|
|
(%, об/об) |
(%, об/об) |
0—25 |
50 → 45 |
50 → 55 |
25—30 |
45 → 40 |
55 → 60 |
30—80 |
40 → 25 |
60 → 75 |
80—81 |
25 → 50 |
75 → 50 |
81—93 |
50 |
50 |
|
|
|
–скорость подвижной фазы: 1,0 мл/мин;
–спектрофотометрический детектор,
длина волны 210 нм;
–объем вводимой пробы: 50 мкл. Относительное удерживание (по отно-
шению к азитромицину; время удерживания — 45—50 мин): примесь L — около 0,29; примесь
М — около 0,37; примесь Е — около 0,43; при-
месь F — около 0,51; примесь D — около 0,54; примесь J — около 0,54; примесь I — около 0,61; примесь C — около 0,73; примесь N —
около 0,76; примесь Н — около 0,79; примесь
А — около 0,83; примесь Р — около 0,92; примесь О — около 1,23; примесь G — около 1,26; примесь В — около 1,31.
Идентификация пиков примесей: идентифицируют пики примесей А, С, Е, F, G, I, J, L, M, N, O и Р, используя хроматограмму, прилага-
емую к ФСО азитромицина для идентификации пиков, и хроматограмму раствора сравнения (с); идентифицируют пик примеси В на хромато-
грамме раствора сравнения (d) и пик примеси Н
на хроматограмме сравнения (b).
Пригодность хроматографической системы: раствор сравнения (b):
–коэффициент разделения пиков: не
менее 1,4 (Hp — высота пика примеси J относи-
тельно базовой линии; Hv — расстояние между
базовой линией и нижней точкой кривой, разде-
ляющей пики примеси J и примеси F).
Предельное содержание примесей (для рас-
чета содержания примесей умножают площади пиков на соответствующие поправочные коэффициенты: для примеси F — 0,3; для примеси Н — 0,1; для примеси L — 2,3; для примеси М — 0,6; для примеси N — 0,7):
–примесь В (не более 2,0%): на хромато-
грамме испытуемого раствора площадь пика
примеси В не должна превышать 2-кратную пло-
щадь основного пика на хроматограмме раст-
вора сравнения (а);
–примеси А, С, Е, F, G, Н, I, L, M, N, O, P (не более 0,5%): на хроматограмме испытуемого
раствора площади пиков, соответствующих примесям А, С, Е, F, G, Н, I, L, M, N, O и P, не должны
превышать 0,5 площади основного пика на хроматограмме раствора сравнения (а);
–сумма примесей D и J (не более 0,5%):
на хроматограмме испытуемого раствора сумма
площадей пиков примесей D и J не должна превышать 0,5 площади основного пика на хроматограмме раствора сравнения (а);
–любая другая примесь (не более 0,2%): на
хроматограмме испытуемого раствора площадь любого пика, кроме основного и пиков примесей
А, B, С, D, Е, F, G, Н, I, J, L, M, N, O и P, не должна
превышать 0,2 площади основного пика на хро-
матограмме раствора сравнения (а);
–сумма примесей (не более 3,0%): на хроматограмме испытуемого раствора сумма площадей всех пиков, кроме основного, не должна превышать 3-кратную площадь основного пика
на хроматограмме раствора сравнения (а).
На хроматограмме испытуемого раствора не учитывают пики с площадью менее 0,1 площади
основного пика на хроматограмме раствора срав-
нения (а) (0,1%) и пики, выходящие перед пиком примеси L и после пика примеси В.
Тяжелые металлы (2.4.8, метод В). Не более 0,0025% (25 ppm). 2,0 г испытуемого образца растворяют в смеси вода Р — этанол Р (15:85, об/об) и
доводят до объема 20 мл этим же растворителем.
12 мл полученного раствора должны выдерживать испытание на тяжелые металлы. Эталон готовят
сиспользованием эталонного раствора свинца (2,5 ppm Pb), полученного путем разведения эталонного раствора свинца (100 ppm Pb) Р смесью вода Р — этанол Р (15:85, об/об).
Вода (2.5.12). Не менее 1,8% и не более
6,5%. Определение проводят из 0,200 г испытуемого образца.
Сульфатная зола (2.4.14, метод А). Не
более 0,2%. Определение проводят из 1,0 г испы-
туемого образца.
# Остаточные количества органических растворителей (2.4.24). Испытуемый образец
должен выдерживать требования статьи (5.4).
# Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Азитромицин в условиях испыта-
ния обладает антимикробным действием. Посев
на питательную среду № 2 проводят из разве-
дения 1:50, посев на питательные среды № 1,
№ 11 и № 8 — из разведения 1:50.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Жидкостная хроматография (2.2.29). Раствор А. Смешивают 60 объемов ацето-
нитрила Р1 и 40 объемов раствора 6,7 г/л дикалия гидрофосфата Р, доведенного до рН 8,0
кислотой фосфорной Р.
130 |
Государственная фармакопея Республики Беларусь |
Испытуемый раствор. 53,0 мг испыту-
емого образца растворяют в 2 мл ацетонитрила Р1 и доводят раствором А до объема
100,0 мл.
Раствор сравнения (a). 53,0 мг ФСО азитромицина растворяют в 2 мл ацетонитрила Р1 и доводят раствором А до объема
100,0 мл.
Раствор сравнения (b). 5 мг испытуемого образца и 5 мг ФСО азитромицина примеси А
растворяют в 0,5 мл ацетонитрила Р1 и до-
водят раствором А до объема 10 мл. Условия хроматографирования:
–колонка длиной 0,25 м и внутренним ди-
аметром 4,6 мм, заполненная винилполимером октадецилсилильным для хроматографии Р с размером частиц 5 мкм;
–температура: 40°С;
–подвижная фаза: 60 объемов ацетонитрила Р1 и 40 объемов раствора 6,7 г/л дикалия гидрофосфата Р, доведенного до рН 11,0
раствором 560 г/л калия гидроксида Р;
–скорость подвижной фазы: 1,0 мл/мин;
–спектрофотометрический детектор, длина волны 210 нм;
–объем вводимой пробы: 10 мкл;
–время хроматографирования: 1,5-крат- ное время удерживания азитромицина.
Время удерживания: азитромицин — около 10 мин.
Пригодность хроматографической системы: раствор сравнения (b):
–разрешение: не менее 3,0 между пиками примеси А и азитромицина.
Содержание С38Н72N2O12 рассчитывают в процентах с учетом содержания азитромицина в ФСО азитромицина.
# КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Определение проводят микробиологическим методом (2.7.2, метод А).
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
Специфицированные примеси: A, B, C, D, E, F, G, H, I, J, L, M, N, O, P.
Другие обнаруживаемые примеси (сле-
дующие вещества, если они присутствуют в значительных количествах, следует опреде-
лять тем или иным испытанием, описанным в
частной статье. Их содержание лимитируется
общим критерием приемлемости для других/
неспецифицированных примесей и/или общей
статьей Субстанции для фармацевтического использования. Вследствие этого нет необходимости идентифицировать эти примеси для доказательства соответствия требованиям. См. также статью 5.10 Контроль примесей в субстанциях для фармацевтического использования): К.
|
|
|
|
R2 |
|
|
|
|
|
H3C |
N |
CH3 |
|
|
|
|
H |
H |
H |
|
|
|
|
H3C |
R1 |
|
|
H3C |
|
|
HO |
HO |
CH3 |
|
|
|
|
|
|
|
|
H3C |
R3 O O |
|
O H |
CH3 O |
H |
|
N |
OH |
|
H |
|
|
|
|
|
O |
|
O |
R6 |
|
|
|
CH3 |
|
|
||
|
OH |
H |
|
|
||
|
|
|
|
|||
|
OR4 |
H R5 |
|
|
||
|
|
|
|
|
||
CH3
А. R1 = OH, R2 = R6 = H, R3 = R4 = R5 = CH3: 6-Деметилазитромицин.
В. R1 = R6 = H, R2 = R3 = R4 = R5 = СH3:
3-O-Дезоксиазитромицин (азитромицин В).
С. R1 = OH, R2 = R3 = R5 = CH3, R4 = R6 = H: 3’’-О-Деметилазитромицин (азитромицин С).
D. R1 = OH, R2 = R3 = R4 = CH3, R5 = СН2ОН,
R6 = Н: 14-Деметил-14-(гидроксиметил)азитро- мицин (азитромицин F).
F. R1 = OH, R2 = R4 = R5 = CH3, R3 = СНО, R6 = Н: 3’-N-Деметил-3’-N-формилазитромицин.
I. R1 = OH, R2 = R4 = R5 = CH3, R3 = R6 = Н: 3’-N-Деметилазитромицин.
O. R1 = OH, R2 = R3 = R4 = R5 = R6 = CH3:
2-Дезэтил-2-пропилазитромицин.
|
CH3 |
|
|
|
H3C |
N |
CH3 |
|
|
H |
H |
H |
|
|
H3C |
OH |
|
|
|
HO |
HO |
CH3 |
OH |
O |
|
|
|
||
O |
CH3 |
H |
|
|
|
CH3 |
|||
H |
O |
CH3 |
|
OCH3 |
H |
|
|||
R2 O |
|
O |
|
|
H |
|
|
|
|
|
|
|
CH3 |
|
R1 |
H CH3 |
|
кладинозил |
|
|
|
|||
|
R1 = кладинозил |
|
|
|
|
|
CH3 |
|
|
|
|
O |
|
|
|
R2 = |
NH2 |
|
|
OH
E. 3’-(N,N-Дидеметил)азитромицин (аминоазитромицин).
R1 = кладинозил
O |
O H3C |
S |
CH3O |
R2 = |
N |
|
|
H3C |
OH |
|
G. 3’-N-Деметил-3’-N-[(4-метилфенил)cуль-
фонил]азитромицин.
R1 = кладинозил
|
O |
O H3C |
|
S |
CH3O |
R2 = |
O |
N |
|
|
|
H3C |
N |
OH |
|
H |
|
|
|
H. 3’-N-Деметил-[[4-(ацетиламино)фенил]cуль-
фонил]-3’-N-деметилазитромицин.