2009.-Byelorussian Pharmacopoeia_Volume 3
.pdf# Дротаверина гидрохлорид |
|
|
|
281 |
|||||||||||
Е. 1-[1-[4-[4-[4-(2-оксо-2,3-дигидро-1Н- |
ют при температуре 100°С в течение 3 мин. По- |
||||||||||||||
бензими-дазол-1-ил)-3,6-дигидропиридин-1(2Н)- |
является зеленое окрашивание. После охлажде- |
||||||||||||||
ил]-1-оксобутил]фенил]-1,2,3,6-тетрагидропиридин- |
ния прибавляют 0,5 мл кислоты азотной раз- |
||||||||||||||
4-ил]-1,3-дигидро-2Н-бензимидазол-2-он. |
|
веденной Р. Появляется коричневато-красное |
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
окрашивание. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
D. Испытуемый образец дает реакцию (а) на |
|||
# ДРОТАВЕРИНА ГИДРОХЛОРИД |
|
хлориды (2.3.1). |
|
|
|||||||||||
|
ИСПЫТАНИЯ |
|
|
||||||||||||
Drotaverini hydrochloridum |
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
Раствор S. 0,5 г испытуемого образца раст- |
||||||
DROTAVERINE HYDROCHLORIDE |
|
||||||||||||||
|
воряют в 50,0 мл воды Р. |
|
|||||||||||||
H3C |
O |
|
|
|
|
|
|
|
|
|
Прозрачность |
(2.2.1). Раствор |
S должен |
||
|
|
|
|
|
|
|
|
|
быть прозрачным. |
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
Цветность (2.2.2, метод II). Окраска раст- |
|||
H3C |
|
|
|
NH |
HCl |
вора S должна быть не интенсивнее эталона |
|||||||||
O |
|||||||||||||||
|
|
|
|
|
|
|
|
G(З)5 или Y(Ж)5. |
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
рН (2.2.3). От 3,5 до 5,5. Измеряют рН раст- |
|||
|
|
|
|
|
|
|
|
|
|
|
|
вора S. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Сопутствующие примеси. Жидкостная |
|||
|
|
|
|
|
|
|
|
|
|
|
|
хроматография (2.2.29). |
|
||
|
|
|
|
|
|
|
|
O |
|
Буферный раствор. 10,88 г натрия ацета- |
|||||
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
та Р растворяют в 500 мл воды Р, прибавляют |
|||
|
|
|
|
|
|
O |
|
|
CH3 |
30 мл кислоты уксусной ледяной Р и доводят |
|||||
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
водой Р до объема 1000,0 мл. |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
Испытуемый раствор. 10 мг испытуемого |
|||
|
|
|
|
|
|
|
CH3 |
|
|||||||
|
|
|
|
|
|
|
|
образца растворяют в 5 мл подвижной фазы. |
|||||||
C24H31NO4 · HCl |
|
|
|
|
|
М.м. 434,0 |
Раствор сравнения (а). 2,5 мл испытуемого |
||||||||
ОПРЕДЕЛЕНИЕ |
|
|
|
|
|
|
|
|
|
раствора доводят подвижной фазой до объема |
|||||
|
|
|
|
|
|
|
|
|
25,0 мл. 0,5 мл полученного раствора доводят |
||||||
Дротаверина гидрохлорид содержит не |
подвижной фазой до объема 10,0 мл. |
||||||||||||||
менее 98,0 % и не более 101,0 % 1-[(3,4-ди- |
Раствор сравнения (b). 20 мг испытуемого |
||||||||||||||
этоксифенил)метилен]-6,7-диэтокси-3,4- |
образца растворяют в 19 мл воды Р при темпе- |
||||||||||||||
дигидро-2Н-изохинолина гидрохлорида в пе- |
ратуре от 40°С до 50°С, прибавляют 5 мг рас- |
||||||||||||||
ресчете на безводное и свободное от этано- |
тертого калия перманганата Р, перемешивают |
||||||||||||||
ла вещество. |
|
|
|
|
|
|
|
|
|
в течение 2 мин, прибавляют 1 мл кислоты фос- |
|||||
ОПИСАНИЕ (СВОЙСТВА) |
|
форной Р и перемешивают до получения про- |
|||||||||||||
|
зрачного раствора. |
|
|
||||||||||||
Кристаллический порошок от светло- |
Условия хроматографирования: |
|
|||||||||||||
желтого до зеленовато-желтого цвета. |
|
– колонка длиной 0,25 м и внутренним диа- |
|||||||||||||
Умеренно растворим в воде, растворим в |
метром 4,6 мм, заполненная силикагелем окта- |
||||||||||||||
96 % спирте, легкорастворим в хлороформе. |
децилсилильным деактивированным по отно- |
||||||||||||||
Температура плавления: от 205°С до |
шению к основаниям эндкепированным для хро- |
||||||||||||||
215°С (без предварительного подсушивания) с |
матографии Р с размером частиц 5 мкм; |
||||||||||||||
разложением. |
|
|
|
|
|
|
|
|
|
– подвижная фаза: метанол Р — ацетони- |
|||||
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ) |
|
трил для хроматографии Р — буферный раст- |
|||||||||||||
|
вор (2:12:11, об/об/об); |
|
|||||||||||||
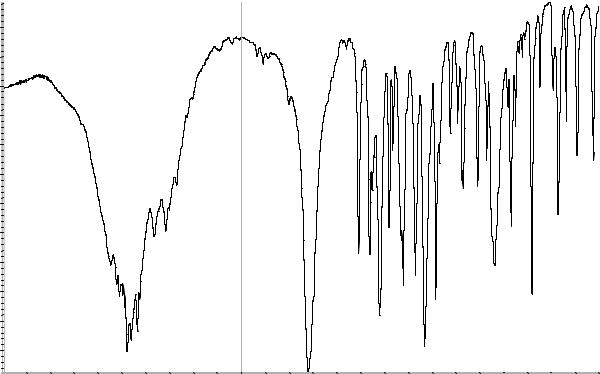
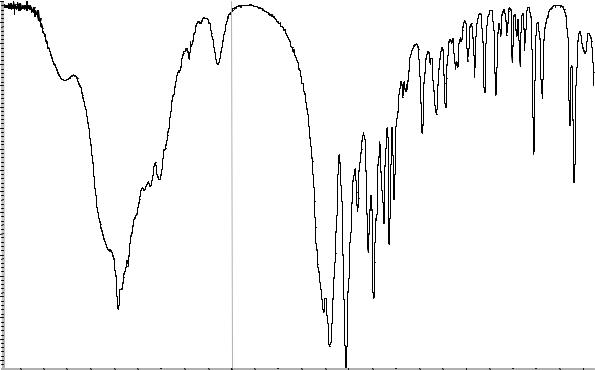
А. Абсорбционная спектрофотометрия в ин- |
– скорость подвижной фазы: 1,0 мл/мин; |
||||||||||||||
фракрасной области (2.2.24). |
|
– спектрофотометрический |
детектор, |
||||||||||||
Сравнение: ФСО дротаверина гидрохлори- |
длина волны 254 нм; |
|
|||||||||||||
да или спектр, представленный на рисунке 1. |
– объем вводимой пробы: по 10 мкл раст- |
||||||||||||||
В. |
Абсорбционная |
|
спектрофотометрия |
вора сравнения (а) и испытуемого раствора и по |
|||||||||||
в ультрафиолетовой и |
|
видимой областях |
5 мкл раствора сравнения (b); |
|
|||||||||||
(2.2.25). 1,5 мг испытуемого образца раство- |
– время хроматографирования: 2,5-крат- |
||||||||||||||
ряют в 0,01 М растворе кислоты хлористово- |
ное время удерживания дротаверина. |
||||||||||||||
дородной и доводят до объема 100,0 мл этим |
Пригодность |
хроматографической сис- |
|||||||||||||
же растворителем. Спектр поглощения полу- |
темы: |
|
|
||||||||||||
ченного раствора в диапазоне длин волн от |
– после пика дротаверина элюируются 3 |
||||||||||||||
220 нм до 400 нм имеет максимумы при 241 нм, |
пика примесей (продукты окисления) на хрома- |
||||||||||||||
302 нм и 353 нм. |
|
|
|
|
|
|
|
|
|
тограмме раствора сравнения (b); |
|
||||
С. 10 мг испытуемого образца растворяют |
– разрешение: не менее 3,0 между со- |
||||||||||||||
в 5 мл кислоты серной Р, прибавляют 0,5 мл |
седними пиками на хроматограмме раствора |
||||||||||||||
раствора 30 г/л железа (III) хлорида Р и нагрева- |
сравнения (b); |
|
|
||||||||||||