

2009.-Byelorussian Pharmacopoeia_Volume 3
.pdfКальция лактат тригидрат |
321 |
вают в течение 4 ч, доводят водой Р до объема
100,0 мл и фильтруют. К 50,0 мл полученного
фильтрата прибавляют 0,5 мл кислоты серной Р, выпаривают досуха и сжигают остаток при температуре (600±50)°С до постоянной массы.
Масса полученного остатка не должна превы-
шать 5 мг.
Тяжелые металлы (2.4.8, метод А). Не
более 0,001 % (10 ppm). Навеску испытуемо-
го образца, соответствующую 2,0 г сухого ве-
щества, растворяют в воде Р и доводят до
объема 20 мл этим же растворителем. 12 мл полученного раствора должны выдерживать
испытание на тяжелые металлы. Эталон готовят с использованием эталонного раствора свинца (1 ррm Pb) Р.
Потеря в массе при высушивании
(2.2.32). Не менее 22,0 % и не более 27,0 %. 0,500 г испытуемого образца сушат при температуре 125°С.
#Остаточные количества органических растворителей (2.4.24). Испытуемый образец
должен выдерживать требования статьи (5.4).
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Кальция лактат пентагидрат в
условиях испытания не обладает антимикроб-
ным действием.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Навеску испытуемого образца, соответству-
ющую 0,200 г сухого вещества, растворяют в
воде Р, доводят до объема 300 мл этим же растворителем и проводят комплексометрическое определение кальция (2.5.11).
1 мл 0,1 М раствора натрия эдетата соот-
ветствует 21,82 мг C6H10CaO6.
КАЛЬЦИЯ ЛАКТАТ ТРИГИДРАТ
Calcii lactas trihydricus
CALCIUM LACTATE TRIHYDRATE
|
|
H C |
CO - |
|
|
Ca2+ |
3 |
2 |
|
и энантиомер 3H2O |
|
|
|
|
|||
|
|
H OH |
|
2 |
|
|
|
|
|
|
|
C6H10CaO6 · 3H2O |
|
|
М.м. 272,3 |
||
ОПРЕДЕЛЕНИЕ
Кальция лактат тригидрат содержит не менее 98,0% и не более 102,0% кальция бис(2- гидроксипропаноата) или смеси кальция (2R)-, (2S)- и (2RS)-2-гидроксипропаноатов в пересчете на сухое вещество.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический либо гранулированный порошок.
Растворим в воде, легкорастворим в кипящей воде, очень мало растворим в 96% спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Испытуемый образец выдерживает испы-
тание «Потеря в массе при высушивании» как указано в разделе «Испытания».
В. Испытуемый образец дает реакцию на лактаты (2.3.1).
С. Испытуемый образец дает реакцию (b) на
кальций (2.3.1).
ИСПЫТАНИЯ
Раствор S. 6,2 г испытуемого образца
(соответствует 5,0 г сухого вещества) раст-
воряют при нагревании в воде, свободной от углерода диоксида, Р, приготовленной из воды дистиллированной Р, охлаждают и до-
водят до объема 100 мл этим же раствори-
телем.
Прозрачность (2.2.1). Раствор S по степени мутности не должен превышать эталон II.
Цветность (2.2.2, метод II). Окраска раствора S должна быть не интенсивнее эта-
лона BY(КЖ)6.
Кислотность или щелочность. К 10 мл
раствора S прибавляют 0,1 мл раствора фенолфталеина Р и 0,5 мл 0,01 М раствора кислоты хлористоводородной. Раствор должен
быть бесцветным. При прибавлении не более
2,0 мл 0,01 М раствора натрия гидроксида
окраска раствора должна измениться на розовую.
Хлориды (2.4.4). Не более 0,02 % (200 ppm). 5 мл раствора S доводят водой Р до объема 15 мл.
Сульфаты (2.4.13). Не более 0,04 % (400 ppm). 7,5 мл раствора S доводят водой дистиллированной Р до объема 15 мл.
Барий. К 10 мл раствора S прибавляют 1 мл раствора кальция сульфата Р и выдерживают в течение 15 мин. Опалесценция полученного раствора не должна превышать опалесценцию смеси из 1 мл воды дистиллированной Р и 10 мл раствора S.
Железо (2.4.9). Не более 0,005 % (50 ppm). 4 мл раствора S доводят водой Р до объема 10 мл.
Соли магния и щелочных металлов. Не более 1 %. К 20 мл раствора S прибавляют 20 мл воды Р, 2 г аммония хлорида Р и 2 мл раствора аммиака разведенного Р1. Нагревают до кипения и быстро прибавляют 40 мл горячего раствора аммония оксалата Р. Выдерживают в течение 4 ч, доводят водой Р до
объема 100,0 мл и фильтруют. К 50,0 мл по-
лученного фильтрата прибавляют 0,5 мл кислоты серной Р, выпаривают досуха и сжигают остаток при температуре (600±50)°С до постоянной массы. Масса полученного остатка не должна превышать 5 мг.
Тяжелые металлы (2.4.8, метод А). Не более 0,001 % (10 ppm). Навеску испытуемого образца, соответствующую 2,0 г сухого ве-
щества, растворяют в воде Р и доводят до
322 |
Государственная фармакопея Республики Беларусь |
объема 20 мл этим же растворителем. 12 мл
полученного раствора должны выдерживать
испытание на тяжелые металлы. Эталон гото-
вят с использованием эталонного раствора свинца (1 ррm Pb) Р.
Потеря в массе при высушивании
(2.2.32). Не менее 15,0 % и не более 20,0 %.
0,500 г испытуемого образца сушат при температуре 125°С.
#Остаточные количества органических растворителей (2.4.24). Испытуемый образец
должен выдерживать требования статьи (5.4).
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Кальция лактат тригидрат в условиях испытания не обладает антимикробным действием.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Навеску испытуемого образца, соответствующую 0,200 г сухого вещества, растворяют в
воде Р, доводят до объема 300 мл этим же раст-
ворителем и проводят комплексометрическое
определение кальция (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответствует 21,82 мг C6H10CaO6.
КАЛЬЦИЯ ФОЛИНАТ
Calcii folinas
CALCIUM FOLINATE
|
|
|
|
|
|
|
|
O |
H CO2- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
CHO |
|
|
|
|
N |
||
|
|
|
|
|
|
|||||
Ca2+ |
|
|
|
|
|
|
|
|
|
H |
|
|
|
N |
|
|
|
|
x H2O |
||
|
|
|
|
|
|
|
||||
|
N |
|
|
N |
CO2- |
|||||
|
|
|
|
|
H |
H |
||||
|
|
|
|
|
|
|||||
H2N |
|
N |
N |
|
|
|
|
и эпимер у С* |
||
|
|
H |
|
|
|
|
|
|
||
С20H21CaN7O7 · хН2О |
|
|
|
|
М.м. 511,5 |
|||||
|
|
|
|
|
|
|
(безводное вещество) |
|||
ОПРЕДЕЛЕНИЕ
Кальция фолинат представляет собой кальция (2S)-2-[[4-[[[(6RS)-2-амино-5-формил-4-оксо-
1,4,5,6,7,8-гексагидроптеридин-6-ил]метил]-
амино]бензоил]амино]пентандиоат.
Содержание:
– кальция фолинат (C20H21CaN7O7): не менее 97,0% и не более 102,0% в пересчете на
безводное вещество;
–кальций (Ca, А.м. 40,08): не менее 7,54%
ине более 8,14% в пересчете на безводное вещество.
ОПИСАНИЕ (СВОЙСТВА)
Белый или светло-желтый аморфный либо кристаллический порошок. Гигроскопичен.
Умеренно растворим в воде, практически нерастворим в ацетоне и в 96% спирте.
Аморфная форма может давать пересыщенные растворы в воде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А, В, D. Вторая идентификация: А, С, D.
А. Испытуемый образец выдерживает испытание «Удельное оптическое вращение» как указано в разделе «Испытания».
В. Абсорбционная спектрофотометрия в ин-
фракрасной области (2.2.24).
Приготовление: в дисках.
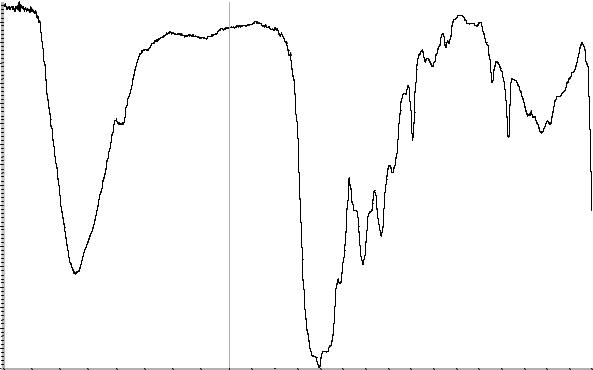
Сравнение: ФСО кальция фолината # или спектр, представленный на рисунке 1.
Если полученные спектры отличаются, то
испытуемый образец и ФСО кальция фолината растворяют по отдельности в минимальном количестве воды Р, прибавляют по каплям до-
статочное количество ацетона Р для получе-
ния осадков и выдерживают в течение 15 мин.
Осадки собирают с помощью центрифугирова-
ния, дважды промывают малым количеством ацетона Р, высушивают и используют для полу-
чения новых спектров.
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 15 мг испытуемого образца растворяют в смеси из 3 объемов раствора аммиака Р и 97 объемов воды Р и доводят
до объема 5 мл этой же смесью растворителей.
Раствор сравнения. 15 мг ФСО кальция фолината растворяют в смеси из 3 объемов раствора аммиака Р и 97 объемов воды Р и доводят до объема 5 мл этой же смесью растворителей.
Пластинка: ТСХ пластинка со слоем целлюлозы для хроматографии F254 Р.
Подвижная фаза: используют нижний слой смеси из 1 объема изоамилового спирта Р и 10 объемов раствора 50 г/л лимонной кислоты Р,
предварительно доведенного до рН 8 раствором аммиака Р.
Наносимый объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от линии старта.
Высушивание: на воздухе.
Проявление: пластинку просматривают в ультрафиолетовом свете при длине волны 254 нм.
Результаты: на хроматограмме испытуе-
мого раствора обнаруживается пятно, соответствующее по расположению и размеру основному пятну на хроматограмме раствора сравнения.
D. Испытуемый образец дает реакцию (b) на кальций (2.3.1).
Испытания и количественное определе-
ние проводят как можно быстрее и с защитой от
света, способного вызвать фотохимические пре-
вращения.
ИСПЫТАНИЯ
Раствор S. 1,25 г испытуемого образца растворяют в воде, свободной от углерода диоксида, Р, при необходимости нагревая до 40°С, и доводят до объема 50,0 мл этим же
растворителем.
Кальция фолинат |
323 |
Прозрачность (2.2.1). Раствор S должен
быть прозрачным.
Оптическая плотность (2.2.25). Не более
0,60 при 420 нм. Измеряют оптическую плотность раствора S, используя в качестве компен-
сационного раствора воду Р.
рН (2.2.3). От 6,8 до 8,0. Измеряют рН раствора S.
Удельное оптическое вращение (2.2.7). От +14,4 до +18,0 в пересчете на безводное вещество. Определяют удельное оптическое вращение раствора S.
Ацетон, этанол и метанол. Газовая хрома-
тография (2.2.28) с использованием парофазного пробоотборника. Используют метод стандарт-
ных добавок.
Испытуемый раствор. 0,25 г испытуемо-
го образца растворяют в воде Р и доводят до объема 10,0 мл этим же растворителем.
Раствор сравнения. 0,125 г ацетона Р, 0,750 г этанола Р и 0,125 г метанола Р раст-
воряют в воде Р и доводят до объема 1000,0 мл
этим же растворителем.
Условия хроматографирования:
–колонка кварцевая капиллярная длиной 10 м и диаметром 0,32 мм, покрытая слоем сополимера стирола-дивинилбензола Р;
–газ-носитель: азот для хроматографии Р;
–скорость газа-носителя: 4 мл/мин;
–параметры парофазного пробоотборника:
–равновесная температура: 80°С,
–время достижения равновесия: 20 мин,
–время приложения избыточного давления: 30 с;
–температура:
|
Время (мин) |
Температура |
|
(°С) |
|
|
|
|
Колонка |
0—6 |
125 → 185 |
|
6—15 |
185 |
|
|
|
Блок ввода |
|
250 |
проб |
|
|
Детектор |
|
250 |
|
|
|
–детектор: пламенно-ионизационный;
–объем вводимой пробы: проводят не
менее 3 вводов.
Предельное содержание:
–ацетон: не более 0,5%;
–этанол: не более 3,0%;
–метанол: не более 0,5%.
Сопутствующие примеси. Жидкостная
хроматография (2.2.29).
Испытуемый раствор. 10,0 мг испытуемого образца растворяют в воде Р и доводят до
объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 10,0 мг ФСО кальция фолината растворяют в воде Р и доводят до объема 10,0 мл этим же растворителем.
Раствор сравнения (b). 1,0 мл раствора
сравнения (а) доводят водой Р до объема
100,0 мл.
Раствор сравнения (с). 10,0 мг ФСО формилфолиевой кислоты (примесь D) растворяют в подвижной фазе и доводят до объема 100,0 мл
этим же растворителем. 1,0 мл полученного раствора доводят водой Р до объема 10,0 мл.
Раствор сравнения (d). 1,0 мл раствора сравнения (b) доводят водой Р до объема 10,0 мл.
Пропускание
95 |
|
|
|
|
|
90 |
|
|
|
|
|
85 |
|
|
|
|
|
80 |
|
|
|
|
|
75 |
|
|
|
|
|
70 |
|
|
|
|
|
65 |
|
|
|
|
|
60 |
|
|
|
|
|
55 |
|
|
|
|
|
50 |
|
|
|
|
|
45 |
|
|
|
|
|
40 |
|
|
|
|
|
35 |
|
|
|
|
|
30 |
|
|
|
|
|
25 |
|
|
|
|
|
20 |
|
|
|
|
|
15 |
|
|
|
|
|
10 |
|
|
|
|
|
4000 |
3000 |
2000 |
1500 |
1000 |
500 |
|
|
Волновое |
число (см-1) |
|
|
Рисунок 1. Инфракрасный спектр пропускания ФСО кальция фолината в дисках с калия бромидом Р.
324 |
Государственная фармакопея Республики Беларусь |
Раствор сравнения (e). 5,0 мл раствора
сравнения (с) доводят раствором сравнения (b)
до объема 10,0 мл.
Условия хроматографирования:
–колонка длиной 0,25 м и внутренним диаметром 4,0 мм, заполненная силикагелем октадецилсилильным для хроматографии Р с раз-
мером частиц 5 мкм;
–температура: 40°С;
–подвижная фаза: смешивают 220 мл метанола Р и 780 мл раствора, содержащего
2,0 мл раствора тетрабутиламмония гидроксида (400 г/л) Р и 2,2 г дикалия гидрофосфата Р, предварительно доведенного до рН 7,8 кислотой фосфорной Р;
–скорость подвижной фазы: 1 мл/мин;
–спектрофотометрический детектор,
длина волны 280 нм;
–объем вводимой пробы: по 10 мкл испытуемого раствора и растворов сравнения (b), (c),
(d)и (е);
–время хроматографирования: 2,5-крат-
ное время удерживания фолината.
Пригодность хроматографической системы: раствор сравнения (е):
— разрешение: не менее 2,2 между пиками
фолината и примеси D.
Предельное содержание примесей:
–примесь D (не более 1%): на хроматограмме испытуемого раствора площадь пика соответствующего примеси D, не должна превы-
шать площадь основного пика на хроматограм-
ме раствора сравнения (с);
–примеси А, В, С, Е, F, G (не более 1%): на хроматограмме испытуемого раствора площадь
любого пика, кроме основного и пика примеси D,
не должна превышать площадь основного пика на хроматограмме раствора сравнения (b);
–сумма примесей, кроме примеси D (не более 2,5%): на хроматограмме испытуемо-
го раствора сумма площадей всех пиков, кроме основного и пика примеси D, не должна превы-
шать 2,5-кратную площадь основного пика на хроматограмме раствора сравнения (b).
На хроматограмме испытуемого раствора не учитывают пики с площадью менее площади основного пика на хроматограмме раствора сравнения (d) (0,1%).
Хлориды. Не более 0,5 %. 0,300 г испыту-
емого образца растворяют в 50 мл воды Р, при
необходимости нагревая до 40°С. К полученно-
му раствору прибавляют 10 мл 2 М раствора кислоты азотной и титруют 0,005 М раствором серебра нитрата потенциометрически
(2.2.20).
1 мл 0,005 М раствора серебра нитрата
соответствует 0,177 мг Cl.
Тяжелые металлы (2.4.8, метод F). 1,0 г испытуемого образца должен выдерживать испытание на тяжелые металлы. Эталон готовят с использованием 5 мл эталонного раствора свинца (10 ppm Pb) Р.
Платина. Не более 0,002% (20,0 ppm).
Атомно-абсорбционная спектрометрия (2.2.23, метод 2).
Испытуемый раствор. 1,00 г испытуемо-
го образца растворяют в воде Р и доводят до объема 100,0 мл этим же растворителем.
Растворы сравнения. Готовят растворы
сравнения с использованием эталонного раствора платины (30 ppm Pt) Р (при необходимости разводят смесью из 1 объема кислоты азотной Р и 99 объемов воды Р).
Источник излучения: лампа с полым катодом для определения платины.
Длина волны: 265,9 нм.
Вода (2.5.12). Не более 17,0%. 0,100 г ис-
пытуемого образца растворяют в смеси из 50 мл
растворителя и 15 мл формамида Р. Перед ти-
трованием смесь перемешивают в течение 6 мин. Используют подходящий титрант, не со-
держащий пиридин.
Бактериальные эндотоксины (2.6.14). Менее 0,5 МЕ/мг, если субстанция предназначена
для производства лекарственных средств для парентерального применения без последующей про-
цедуры удаления бактериальных эндотоксинов.
#Стерильность (2.6.1). Если субстанция предназначена для производства лекарственных средств для парентерального применения без до-
полнительной процедуры стерилизации, то она
должна выдерживать испытание на стерильность.
#Остаточные количества органических растворителей (2.4.24). Испытуемый образец должен выдерживать требования статьи
(5.4). Испытание проводят для растворите-
лей, использующихся в процессе производства субстанции и не перечисленных в испытании «Ацетон, этанол и метанол».
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Кальция фолинат в условиях испы-
тания не обладает антимикробным действием.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Кальций. 0,400 г испытуемого образца раст-
воряют в 150 мл воды Р, доводят до объема 300 мл
этим же растворителем и проводят комплексометрическое определение кальция (2.5.11).
1 мл 0,1 М раствора натрия эдетата соответствует 4,008 мг Ca.
Кальция фолинат. Жидкостная хроматография (2.2.29), как указано в испытании «Сопутствующие примеси», со следующими изменениями.
Объем вводимой пробы: по 5 мкл испытуемого раствора и раствора сравнения (а).
Пригодность хроматографической системы: раствор сравнения (а):
– относительное стандартное отклонение: не более 2,0% для площади основного пика при шести повторных вводах пробы.
Содержание С20H21CaN7O7 рассчитывают с учетом содержания кальция фолината в ФСО кальция фолината.

Каолин тяжелый (# глина белая) |
325 |
ХРАНЕНИЕ
В воздухонепроницаемом контейнере в защищенном от света месте.
Стерильная субстанция — в стерильном воздухонепроницаемом контейнере с контролем первого вскрытия.
ПРИМЕСИ
Специфицированные примеси: А, В, С, D, E, F, G.
OH CO2H
N
H
H2N |
CO2H |
А. (2S)-2-[(4-Аминобензоил)амино]пентан- дионовая кислота.
|
|
|
O |
H CO2H |
|
O |
CHO |
|
N |
|
|
N |
|
H |
|
N |
N |
CO2H |
|
|
|
|||
|
|
|
H |
|
H2N |
N |
N |
CHO |
и эпимер у С* |
|
||||
|
H |
|
|
|
В. (2S)-2-[[4-[[[(6RS)-2-Амино-5-формил-4- оксо-1,4,5,6,7,8-гексагидроптеридин-6-ил]метил]фор- миламино]бензоил]амино]пентандионовая кислота (5,10-диформилтетрагидрофолиевая кислота).
С. Фолиевая кислота.
|
|
O |
H CO2H |
|
O |
|
N |
|
|
N |
H |
|
|
|
|
|
N |
N |
CO2H |
|
|
CHO |
|
H2N |
N |
N |
|
|
H |
|
|
D. (2S)-2-[[4-[[(2-Амино-4-оксо-1,4-дигидро- птеридин-6-ил)метил]формиламино]бензоил]- амино]пентандионовая кислота (10-формилфо- лиевая кислота).
CO2H
OCHO
|
N |
N |
N |
|
|
||
|
|
H |
H |
H2N |
N |
N |
и энантиомер |
|
H |
H |
|
Е. 4-[[[(6RS)-2-Амино-5-формил-4-оксо- 1,4,5,6,7,8-гексагидроптеридин-6-ил]метил]фор- миламино]бензоил]амино]бензойная кислота (5-формилтетрагидроптероевая кислота).
|
|
O |
H CO2H |
|
O |
|
N |
|
|
N |
H |
|
|
|
|
|
N |
N |
CO2H |
|
|
R |
|
H2N |
N |
N |
|
|
H |
H |
|
F. R = СНО: (2S)-2-[[4-[[(2-Амино-4-оксо- 1,4,7,8-тетрагидроптеридин-6-ил)метил]форми-
ламино]бензоил]амино]пентандионовая кислота (10-формилдигидрофолиевая кислота).
G. R = Н: (2S)-2-[[4-[[(2-Амино-4-оксо-1,4,7,8- тетрагидроптеридин-6-ил)метил]амино]бензоил]- амино]пентандионовая кислота (дигидрофолиевая кислота).
КАОЛИН ТЯЖЕЛЫЙ (# ГЛИНА БЕЛАЯ)
Kaolinum ponderosum (# Bolus alba)
KAOLIN, HEAVY
ОПРЕДЕЛЕНИЕ
Каолин тяжелый представляет собой очи-
щенный природный гидратированный силикат
алюминия различного состава.
ОПИСАНИЕ (СВОЙСТВА)
Мелкий белый или серовато-белый мас-
лянистый на ощупь порошок.
Практически нерастворим в воде и в органических растворителях.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 0,5 г испытуемого образца помещают в металлический тигель, прибавляют 1 г калия нитрата Р, 3 г натрия карбоната Р и нагревают до расплавления смеси. Охлажда-
ют, к остатку прибавляют 20 мл кипящей воды Р, перемешивают, фильтруют и промывают
остаток 50 мл воды Р. К остатку прибавляют 1 мл кислоты хлористоводородной Р, 5 мл
воды Р и фильтруют. К фильтрату прибавляют 1 мл раствора натрия гидроксида концентрированного Р и фильтруют. К полученному фильтрату прибавляют 3 мл раствора аммония хлорида Р. Образуется белый гелеобразный осадок.
В. В мерный цилиндр вместимостью 100 мл и диаметром около 30 мм помещают
100 мл раствора 10 г/л натрия лаурилсульфата Р и прибавляют 2,0 г испытуемого образца двадцатью порциями. После прибавления
каждой порции испытуемого образца раствор
выдерживают в течение 2 мин для оседания частиц. Выдерживают в течение 2 ч. Кажущийся объем осадка должен быть не более 5 мл.
С. 0,25 г испытуемого образца дают реакцию на силикаты (2.3.1).
ИСПЫТАНИЯ
Раствор S. К 4 г испытуемого образца
прибавляют смесь из 6 мл кислоты уксусной Р и 34 мл воды дистиллированной Р, встряхивают в течение 1 мин и фильтруют.
Кислотность или щелочность. К 1,0 г испытуемого образца прибавляют 20 мл воды, свободной от углерода диоксида, Р, встряхивают в течение 2 мин и фильтруют. К 10 мл полученного фильтрата прибавляют 0,1 мл раствора фенолфталеина Р. Раствор должен быть бесцветным. При прибавлении не более 0,25 мл
0,01 М раствора натрия гидроксида окраска
раствора должна измениться на розовую.
Органические примеси. 0,3 г испытуемого образца нагревают до красного каления. Полученный остаток может быть лишь незначительно больше окрашен, чем исходный испытуемый образец.

326 |
Государственная фармакопея Республики Беларусь |
Адсорбционная способность. 1,0 г ис-
пытуемого образца помещают в пробирку со
шлифом, прибавляют 10,0 мл раствора 3,7 г/л
метиленового синего Р, встряхивают в течение 2 мин и выдерживают до оседания частиц.
Центрифугируют и разводят водой Р в 100 раз.
Окраска полученного раствора должна быть не интенсивнее окраски раствора 0,03 г/л метиленового синего Р.
Способность к набуханию. 2 г испытуемо-
го образца растирают с 2 мл воды Р. Полученная
смесь не должна быть текучей.
Вещества, растворимые в минеральных кислотах. Не более 1%. К 5,0 г испытуемого образца прибавляют 7,5 мл кислоты хлористоводородной разведенной Р, 27,5 мл воды Р и кипя-
тят в течение 5 мин. Фильтруют, остаток на фильтре промывают водой Р. Фильтрат и промывные воды доводят водой Р до объема 50,0 мл.
К 10,0 мл полученного раствора прибавляют
1,5 мл кислоты серной разведенной Р, выпари-
вают досуха на водяной бане и сжигают. Масса
полученного остатка не должна превышать 10 мг.
Хлориды (2.4.4). Не более 0,025% (250 ppm). 2 мл раствора S доводят водой Р до
объема 15 мл.
Сульфаты (2.4.13). Не более 0,1%. 1,5 мл раствора S доводят водой дистиллированной Р
до объема 15 мл.
Кальций (2.4.3). Не более 0,025% (250 ppm).
4 мл раствора S доводят водой дистиллированной Р до объема 15 мл.
Тяжелые металлы (2.4.8, метод А). Не более 0,005% (50 ppm). К 5 мл раствора, приготовленного как указано в испытании «Вещества,
растворимые в минеральных кислотах», прибав-
ляют 5 мл воды Р, 10 мл кислоты хлористоводородной Р, 25 мл метилизобутилкетона Р
и встряхивают в течение 2 мин. Слои разделяют; водный слой выпаривают на водяной бане
досуха. Полученный остаток растворяют в 1 мл
кислоты уксусной Р, доводят водой Р до объема
25 мл и фильтруют. 12 мл полученного раствора должны выдерживать испытание на тяжелые металлы. Эталон готовят с использованием эталонного раствора свинца (1 ppm Pb) Р.
Если субстанция предназначена для вну-
треннего применения, определение тяжелых ме-
таллов проводят следующим методом (2.4.8, метод А). Не более 0,0025% (25 ppm). К 10 мл
раствора, приготовленного как указано в испы-
тании «Вещества, растворимые в минеральных
кислотах», прибавляют 10 мл воды Р, 20 мл кислоты хлористоводородной Р, 25 мл метилизобутилкетона Р и встряхивают в течение 2 мин. Слои разделяют; водный слой выпаривают на водяной бане досуха. Полученный остаток растворяют в 1 мл кислоты уксусной Р, доводят водой Р
до объема 25 мл и фильтруют. 12 мл полученного раствора должны выдерживать испытание на тяжелые металлы. Эталон готовят с использованием эталонного раствора свинца (1 ppm Pb) Р.
#Остаточные количества органических растворителей (2.4.24). Испытуемый образец
должен выдерживать требования статьи (5.4).
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Каолин тяжелый в условиях испытания не обладает антимикробным действием.
МАРКИРОВКА
При необходимости указывают:
– субстанция пригодна для внутреннего при-
менения.
КАПТОПРИЛ
Captoprilum |
|
|
|
|
|
CAPTOPRIL |
|
|
|
|
|
|
O |
H |
CO2H |
||
|
|
|
|
||
|
|
|
|
|
|
HS |
|
|
|
N |
|
|
|
|
|
||
|
H CH3 |
|
|
||
С9Н15NO3S |
|
|
|
|
М.м. 217,3 |
ОПРЕДЕЛЕНИЕ |
|
|
|||
Каптоприл |
содержит не менее 98,0 % |
||||
и не более |
101,5 % |
(2S)-1-[(2S)-2-метил- |
|||
3 - сульфанилпропаноил]пирролидин - 2 - карбоновой кислоты в пересчете на сухое ве-
щество.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Легкорастворим в воде, в метиленхлори-
де и в метаноле. Растворяется в разведенных
растворах гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфракрасной области (2.2.24).
Сравнение: ФСО каптоприла # или спектр, представленный на рисунке 1.
ИСПЫТАНИЯ
Раствор S. 0,5 г испытуемого образца растворяют в воде, свободной от углерода диоксида, Р и доводят до объема 25,0 мл этим же растворителем.
Прозрачность (2.2.1). Раствор S должен быть прозрачным.
Цветность (2.2.2, метод II). Раствор S должен быть бесцветным.
рН (2.2.3). От 2,0 до 2,6. Измеряют
рН раствора S.
Удельное оптическое вращение (2.2.7). От -127 до -132 в пересчете на сухое вещество. 0,250 г испытуемого образца растворяют в этаноле Р и доводят до объема 25,0 мл этим же растворителем.

Каптоприл |
327 |
Пропускание
98
96
94
92
90
88
86
84
82
80
78
76
74
72
70
3000 |
2000 |
1500 |
1000 |
|
Волновое |
число (см-1) |
|
Рисунок 1. Инфракрасный спектр пропускания ФСО каптоприла. |
|
||
Сопутствующие примеси. Жидкостная |
Предельное содержание примесей: |
||
хроматография (2.2.29). |
|
– любая примесь (не более 1,0%): на хро- |
|
Испытуемый раствор. 50 мг испытуемо- |
матограмме испытуемого раствора |
площадь |
|
го образца растворяют в подвижной фазе и |
любой примеси не должна превышать 0,5 пло- |
||
доводят до объема 100,0 мл этим же раство- |
щади основного пика на хроматограмме раст- |
||
рителем. |
|
вора сравнения (а); |
|
Раствор сравнения (a). 2,0 мл испытуе- |
– сумма примесей (не более 2,0%): на хро- |
||
мого раствора доводят подвижной фазой до |
матограмме испытуемого раствора сумма пло- |
||
объема 100,0 мл. |
|
щадей всех пиков, кроме основного, не должна |
|
Раствор сравнения (b). 10 мг испытуемо- |
превышать площадь основного пика на хромато- |
||
го образца растворяют в подвижной фазе, при- |
грамме раствора сравнения (а). |
|
|
бавляют 0,25 мл 0,05 М раствора йода и до- |
На хроматограмме испытуемого |
раствора |
|
водят подвижной фазой до объема 100,0 мл. |
не учитывают пики с площадью менее 0,1 пло- |
||
10,0 мл полученного раствора доводят под- |
щади основного пика на хроматограмме раст- |
||
вижной фазой до объема 100,0 мл. |
вора сравнения (а) (0,2%) и пики с временем |
||
Условия хроматографирования: |
удерживания менее 1,4 мин. |
|
|
– колонка длиной 0,125 м и внутренним диа- |
Тяжелые металлы (2.4.8, метод С). Не |
||
метром 4 мм, заполненная силикагелем октил- |
более 0,002% (20 ppm). 1,0 г испытуемого образ- |
||
силильным для хроматографии Р с размером |
ца должен выдерживать испытания на тяжелые |
||
частиц 5 мкм; |
|
металлы. Эталон готовят с использованием 2 мл |
|
– подвижная фаза: кислота |
фосфорная |
эталонного раствора свинца (10 ppm Pb) Р. |
|
Р — метанол Р — вода Р (0,05:50:50, об/об/об); |
Потеря в массе при высушивании (2.2.32). |
||
– скорость подвижной фазы: 1 мл/мин; |
Не более 1,0%. 1,000 г испытуемого образца |
||
– спектрофотометрический |
детектор, |
сушат в вакууме при температуре 60°С в тече- |
|
длина волны 220 нм; |
|
ние 3 ч. |
|
– объем вводимой пробы: 20 мкл; |
Сульфатная зола (2.4.14, метод А). Не |
||
– время хроматографирования: 3-кратное |
более 0,2%. Определение проводят из 1,0 г ис- |
||
время удерживания каптоприла. |
|
пытуемого образца. |
|
Пригодность хроматографической си- |
# Остаточные количества органических |
||
стемы: раствор сравнения (b): |
|
растворителей (2.4.24). Испытуемый образец |
|
– на хроматограмме обнаруживаются 3 пика; |
должен выдерживать требования статьи (5.4). |
||
– разрешение: не менее 2,0 между дву- |
# Микробиологическая чистота |
(2.6.12, |
|
мя основными пиками, элюирующимися по- |
2.6.13, 5.1.4). Каптоприл в условиях испытания |
||
следними. |
|
обладает антимикробным действием. Посев на |
|

328 |
Государственная фармакопея Республики Беларусь |
питательную среду № 1 проводят из разведения
1:50, на питательную среду № 2 — из разведе-
ния 1:20, на питательные среды № 8 и № 11 —
из разведения 1:10.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,150 г испытуемого образца растворяют в
30 мл воды Р и титруют 0,05 М раствором йода
потенциометрически (2.2.20), используя комбинированный платиновый электрод.
1 мл 0,05 М раствора йода соответствует
21,73 мг С9Н15NO3S.
ХРАНЕНИЕ
В воздухонепроницаемом контейнере.
ПРИМЕСИ
HO2C |
H O |
|
O H |
CO2H |
|
|
|
||
|
N |
S S |
N |
|
|
H |
CH3 |
H CH3 |
|
А. (2S,2’S)-1,1’-[Дисульфандиилбис[(2S)-2-
метил-1-оксопропан-3,1-диил]-бис[пирролидин-
2-карбоновая]кислота (каптоприл-дисульфид).
КАРВЕДИЛОЛ
Carvedilolum
CARVEDILOL
HOH
O |
H |
N |
|
|
O |
|
OCH3 |
HN |
|
|
и энантиомер |
C24H26N204 |
М.м. 406,5 |
ОПРЕДЕЛЕНИЕ
Карведилол содержит не менее 99,0% и не более 101,0% (2RS)-1-(9H-карбазол-4-илокси)- 3-[[2-(2-метоксифенокси)этил]амино]пропан-2- oла в пересчете на сухое вещество.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, малорастворим в 96% спирте, практически нерастворим в разведенных кислотах.
Обладает полиморфизмом (5.9).
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Абсорбционная спектрофотометрия в инфракрасной области (2.2.24).
Сравнение: эталонный спектр крведилола по Европейской Фармакопее # или спектр, представленный на рисунке 1.
Если полученные спектры отличаются, то испытуемый образец растворяют в 2-пропано- ле Р, выпаривают до сухого остатка, который ис-
пользуют для получения нового спектра.
ИСПЫТАНИЯ
Сопутствующие примеси. Жидкостная
хроматография (2.2.29).
Испытуемый раствор. 25,0 мг испытуемого образца растворяют в подвижной фазе и доводят до объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 1,0 мл испытуемого раствора доводят подвижной фазой до объема
100,0 мл. 1,0 мл полученного раствора доводят подвижной фазой до объема 10,0 мл.
Раствор сравнения (b). 5,0 мг ФСО карведилола примеси С растворяют в 5,0 мл испыту-
емого раствора и доводят подвижной фазой до
объема 100,0 мл.
Раствор сравнения (с). 1,0 мл раствора
сравнения (b) доводят подвижной фазой до объема 100,0 мл. 2,0 мл полученного раствора
доводят подвижной фазой до объема 10,0 мл.
Условия хроматографирования:
–колонка длиной 0,125 м и внутренним диаметром 4,6 мм, заполненная силикагелем октилсилильным для хроматографии Р с раз-
мером частиц 5 мкм;
–температура: 55°С;
–подвижная фаза: 1,77 г калия дигидрофосфата Р растворяют в воде Р, разводят водой Р
до объема 650 мл, доводят до рН 2,0 кислотой фосфорной Р и прибавляют 350 мл ацетонитрила Р.
–скорость подвижной фазы: 1,0 мл/мин.
–спектрофотометрический детектор,
длина волны 240 нм;
–объем вводимой пробы: 20 мкл;
–время хроматографирования: 8-кратное время удерживания карведилола.
Относительное удерживание (по отношению к карведилолу; время удерживания — около 4 мин): примесь А — около 0,6; примесь С — около 3,5; примесь В — около 6,7.
Пригодность хроматографической системы: раствор сравнения (b):
–разрешение: не менее 17 между пиками карведилола и примеси С;
Предельное содержание примесей (для расчета содержания примесей умножают площади пиков на соответствующие поправочные коэффициенты: для примеси А — 2):
–примесь А (не более 0,2%): на хроматограмме испытуемого раствора площадь пика, соответствующего примеси А, не должна превышать 2-кратную площадь основного пика на хро-
матограмме раствора сравнения (а);
–примесь С (не более 0,02%): на хроматограмме испытуемого раствора площадь пика, соответствующего примеси С, не должна превышать 2-кратную площадь соответствующего пика на хроматограмме раствора сравнения (с);

Карведилол |
329 |
Пропускание
90
85
80
75
70
65
60
55
50
45
40
35
3000 |
2000 |
1500 |
1000 |
|
Волновое |
число (см-1) |
|
Рисунок 1. Инфракрасный спектр пропускания карведилола.
–любая другая примесь (не более 0,1 %): на хроматограмме испытуемого раствора пло-
щадь любого пика, кроме основного и пиков примесей А и С, не должна превышать пло-
щадь основного пика на хроматограмме раствора сравнения (а);
–сумма примесей (не более 0,5%): на хроматограмме испытуемого раствора сумма площадей всех пиков, кроме основного, не должна превышать 5-кратную площадь основного пика на хроматограмме раствора сравнения (а).
На хроматограмме испытуемого раствора не учитывают пики с площадью менее площади основного пика на хроматограмме раствора сравнения (с) (0,01%).
Тяжелые металлы (2.4.8, метод С). Не более 0,001% (10 ppm). 2,0 г испытуемого образца должны выдерживать испытание на тяжелые металлы. Эталон готовят с использованием 2,0 мл эталонного раствора свинца (10 ppm Pb) Р.
Потеря в массе при высушивании (2.2.32). Не более 0,5%. 1,000 г испытуемого образца
сушат при температуре 105°С.
Сульфатная зола (2.4.14, метод А). Не
более 0,1%. Определение проводят из 1,0 г ис-
пытуемого образца.
# Остаточные количества органических растворителей (2.4.24). Испытуемый образец должен выдерживать требования статьи (5.4).
# Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Карведилол в условиях испытаний
не обладает антимикробным действием.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,350 г испытуемого образца растворяют в
60 мл кислоты уксусной безводной Р и титруют
0,1 М раствором кислоты хлорной потенциометрически (2.2.20).
1 мл 0,1 М раствора кислоты хлорной соот-
ветствует 40,65 мг C24H26N2O4.
ПРИМЕСИ
H3CO |
|
OH |
|
|
|
H |
|
|
O |
|
|
O |
|
N |
|
|
O |
||
|
|
||
|
|
|
OCH3 |
HN |
N |
|
|
|
|
|
|
|
HO |
|
|
А.1-[[9-[2-Гидрокси-3-[[2-(2-метоксифенокси)- этил]aмино]пропил]-9H-карбазол-4-ил]oкси]-3- [[2-(2-метоксифенокси)этил]амино]пропан-2-oл.
|
|
OH |
|
|
|
|
|
O |
NH |
|
OH |
|
||
|
|
|
||
|
|
|
|
|
O |
|
N |
O |
|
|
|
|
||
|
|
|
|
|
|
|
|
|
OCH3 |
HN

B. 1,1′-[[2-(2-Метоксифенокси)этил]нитрило]- бис[3-(9H-карбазол-4-илокси)пропан-2-ол].

330 |
Государственная фармакопея Республики Беларусь |
H |
OH |
|
O |
N |
и энантиомер |
|
|
O |
|
|
OCH3 |
HN

C. (2RS)-1-[Бензил[2-(2-метоксифенокси)этил]- aмино]-3-(9H-карбазол-4-илокси)пропан-2-oл.
КЕТОКОНАЗОЛ
Ketoconazolum
KETOCONAZOLE
|
|
Cl |
Cl |
|
|
O |
|
O |
|
O |
|
|
|
|
|
N |
N |
O H |
N |
H3C |
|
|
|
|
|
и энантиомер |
N |
|
|
|
|
C26H28Сl2N4O4 |
|
М. м. 531,4 |
|
ОПРЕДЕЛЕНИЕ
Кетоконазол содержит не менее 99,0 % и не более 101,0 % 1-ацетил-4-[4-[[(2RS,4SR)-2-(2,4-
дихлорфенил)-2-(1Н-имидазол-1-илметил)-1,3- диоксолан-4-ил]метокси]фенил]пиперазина в
пересчете на сухое вещество.
ОПИСАНИЕ (СВОЙСТВА)
Белый или почти белый порошок.
Практически нерастворим в воде, легко-
растворим в метиленхлориде, растворим в метаноле, умеренно растворим в 96 % спирте.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: В. Вторая идентификация: A, С, D.
А. Температура плавления (2.2.14): от
148°С до 152°С.
В. Абсорбционная спектрофотометрия в инфракрасной области (2.2.24).
Приготовление: в дисках.
Сравнение: ФСО кетоконазола # или спектр, представленный на рисунке 1.
С. Тонкослойная хроматография (2.2.27).
Испытуемый раствор. 30 мг испытуемого образца растворяют в подвижной фазе и доводят до объема 5 мл этим же раствори-
телем.
Раствор сравнения (а). 30 мг ФСО кетоконазола растворяют в подвижной фазе и доводят до объема 5 мл этим же растворителем.
Раствор сравнения (b). 30 мг ФСО кетоконазола и 30 мг ФСО эконазола нитрата
растворяют в подвижной фазе и доводят до
объема 5 мл этим же растворителем.
Пластинка: ТСХ пластинка со слоем силикагеля октадецилсилильного Р.
Подвижная фаза: раствор аммония ацетата Р — диоксан Р — метанол Р (20:40:40, об/об/об).
Наносимой объем пробы: 5 мкл.
Фронт подвижной фазы: не менее 15 см от линии старта.
Высушивание: в потоке теплого воздуха.
Проявление: пластинку выдерживают в парах йода до появления пятен и просматри-
вают при дневном свете.
Пригодность хроматографической системы: раствор сравнения (b):
– на хроматограмме обнаруживаются два
полностью разделенных пятна. Результаты: на хроматограмме испыту-
емого раствора обнаруживается пятно, соот-
ветствующее по расположению, цвету и раз-
меру основному пятну на хроматограмме раствора сравнения (а).
D. 30 мг испытуемого образца помещают в фарфоровый тигель, прибавляют 0,3 г
натрия карбоната безводного Р и нагрева-
ют на открытом пламени в течение 10 мин. Охлаждают, полученный остаток растворяют в 5 мл кислоты азотной разведенной Р и фильтруют. К 1 мл фильтрата прибавляют 1 мл воды Р. Полученный раствор дает реак-
цию (а) на хлориды (2.3.1).
ИСПЫТАНИЯ
Раствор S. 1,0 г испытуемого образца растворяют в метиленхлориде Р и доводят
до объема 10 мл этим же растворителем. Прозрачность (2.2.1). Раствор S должен
быть прозрачным.
Цветность (2.2.2, метод II). Окраска раствора S должна быть не интенсивнее эталона BY(КЖ)4.
Угол оптического вращения (2.2.7). От -0,10° до +0,10°. Измеряют угол оптического вращения раствора S.
Сопутствующие примеси. Жидкостная
хроматография (2.2.29).
Испытуемый раствор. 0,100 г испытуемого образца растворяют в метаноле Р и доводят до объема 10,0 мл этим же растворителем.
Раствор сравнения (а). 2,5 мг ФСО кетоконазола и 2,5 мг ФСО лоперамида гидрохлорида растворяют в метаноле Р и доводят до объема 50,0 мл этим же растворителем.
Раствор сравнения (b). 5,0 мл испытуемо-
го раствора доводят метанолом Р до объема 100,0 мл. 1,0 мл полученного раствора доводят метанолом Р до объема 10,0 мл.
Условия хроматографирования:
– колонка длиной 0,10 м и внутренним диаметром 4,6 мм, заполненная силикагелем