

2009.-Byelorussian Pharmacopoeia_Volume 3
.pdf


296 |
Государственная фармакопея Республики Беларусь |
ИНДАПАМИД |
Фронт подвижной фазы: не менее 15 см от |
Indapamidum |
линии старта. |
Высушивание: на воздухе. |
|
INDAPAMIDE |
Проявление: пластинку просматривают |
|
|
|
|
|
|
|
CH3 |
|
в ультрафиолетовом свете при длине волны |
||
|
|
|
|
|
|
|
|
254 нм. |
|
|
|
O |
O |
|
O |
H |
|
Пригодность хроматографической систе- |
|||||
|
|
|
мы: раствор сравнения (b): |
|
|||||||
|
|
|
|
|
и энантиомер |
|
|||||
S |
|
|
|
|
|
|
N |
|
|||
|
|
|
|
|
|
– на хроматограмме обнаруживаются два |
|||||
|
|
|
|
|
|
||||||
H2N |
|
|
|
|
|
|
N |
|
полностью разделенных пятна. |
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
H |
|
|
||
Cl |
|
|
|
|
|
|
|
|
Результаты: на хроматограмме испытуе- |
||
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
мого раствора обнаруживается пятно, соответ- |
|||
C16H16ClN3O3S |
|
М.м. 365,8 |
ствующее по расположению и размеру основ- |
||||||||
ОПРЕДЕЛЕНИЕ |
|
|
ному пятну на хроматограмме раствора сравне- |
||||||||
|
|
ния (а). |
|
|
|||||||
Индапамид содержит не менее 98,0% и |
ИСПЫТАНИЯ |
|
|
||||||||
не более |
102,0% |
4-хлор-N-[(2RS)-2-метил- |
|
|
|||||||
2,3-дигидро-1Н-индол-1-ил]-3-сульфамоил- |
Угол оптического |
вращения |
(2.2.7). От |
||||||||
бензамида в пересчете на безводное вещество. |
-0,02° до +0,02°. 0,250 г испытуемого образца |
||||||||||
ОПИСАНИЕ (СВОЙСТВА) |
|
растворяют в этаноле Р и доводят до объема |
|||||||||
|
25,0 мл этим же растворителем. |
|
|||||||||
Белый или почти белый порошок. |
Сопутствующие |
примеси. |
Жидкостная |
||||||||
Практически нерастворим в воде, раство- |
хроматография (2.2.29) как указано в разделе |
||||||||||
рим в 96% спирте. |
|
|
«Количественное определение». |
|
|||||||
|
|
|
|
|
|
|
|
|
Чувствительность |
системы |
регулируют |
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ) |
таким образом, чтобы высота основного пика на |
||||||||||
Первая идентификация: В. |
|
хроматограмме раствора сравнения (b) состав- |
|||||||||
|
ляла не менее 15% шкалы регистрирующего |
||||||||||
Вторая идентификация: A, С. |
|
||||||||||
|
устройства. |
|
|
||||||||
А. |
|
|
|
|
|
|
|
|
|
|
|
Абсорбционная спектрофотометрия в |
Объем вводимой пробы: по 10 мкл каждого |
||||||||||
ультрафиолетовой и видимой областях (2.2.25). |
раствора. |
|
|
||||||||
Испытуемый раствор. 50,0 мг испытуемо- |
Время хроматографирования: 2,5-кратное |
||||||||||
го образца растворяют в 96% спирте Р и дово- |
время удерживания основного пика. |
|
|||||||||
дят до объема 100,0 мл этим же растворителем. |
Пригодность хроматографической сис- |
||||||||||
2,0 мл полученного раствора доводят 96% спир- |
темы: |
|
|
||||||||
том Р до объема 100,0 мл. |
|
– разрешение: не менее 4,0 между пиками |
|||||||||
Диапазон длин волн: от 220 нм до 350 нм. |
индапамида и метилнитрозоиндолина на хрома- |
||||||||||
Максимум поглощения: при 242 нм. |
тограмме раствора сравнения (d); |
|
|||||||||
Плечи: при 279 нм и при 287 нм. |
|
– отношение сигнал/шум: не менее 6 для |
|||||||||
Удельный показатель поглощения в макси- |
основного пика на хроматограмме раствора |
||||||||||
муме: от 590 до 630. |
|
|
сравнения (b). |
|
|
||||||
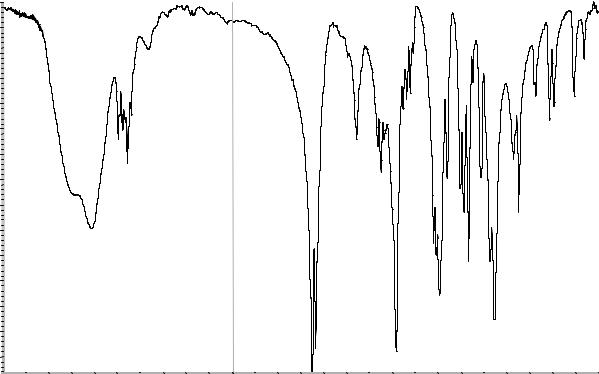
В. Абсорбционная спектрофотометрия в ин- |
Предельное содержание примесей: |
||||||||||
фракрасной области (2.2.24). |
|
– примесь В (не более 0,3%): на хромато- |
|||||||||
Приготовление: в дисках. |
|
грамме испытуемого раствора площадь пика |
|||||||||
Сравнение: ФСО индапамида # или спектр, |
примеси В не должна превышать площадь |
||||||||||
представленный на рисунке 1. |
|
основного пика на хроматограмме раствора |
|||||||||
С. Тонкослойная хроматография (2.2.27). |
сравнения (а); |
|
|
||||||||
Испытуемый раствор. 20 мг испытуе- |
– любая другая примесь (не более 0,1 %): |
||||||||||
мого образца растворяют в 96 % спирте Р |
на хроматограмме испытуемого раствора пло- |
||||||||||
и доводят до объема 10 мл этим же раство- |
щадь любого пика, кроме основного и пика |
||||||||||
рителем. |
|
|
|
|
|
|
примеси В, не должна превышать площадь |
||||
Раствор сравнения (а). 20 мг ФСО индапа- |
основного пика на хроматограмме раствора |
||||||||||
мида растворяют в 96% спирте Р и доводят до |
сравнения (b); |
|
|
||||||||
объема 10 мл этим же растворителем. |
– сумма примесей (не более 0,5 %): на хро- |
||||||||||
Раствор сравнения (b). 10 мг ФСО индоме- |
матограмме испытуемого раствора сумма пло- |
||||||||||
тацина растворяют в 5 мл раствора сравнения |
щадей всех пиков, кроме основного, не должна |
||||||||||
(а) и доводят 96% спиртом Р до объема 10 мл. |
превышать 5-кратную площадь основного пика |
||||||||||
Пластинка: ТСХ пластинка со слоем сили- |
на хроматограмме раствора сравнения (b). |
||||||||||
кагеля GF254 Р. |
|
|
На хроматограмме испытуемого раствора |
||||||||
Подвижная фаза: кислота уксусная ледяная |
не учитывают пики с площадью менее 0,5 пло- |
||||||||||
Р — ацетон Р — толуол Р (1:20:79, об/об/об). |
щади основного пика на хроматограмме раст- |
||||||||||
Наносимый объем пробы: 10 мкл. |
вора сравнения (b) (0,05 %). |
|
|||||||||



 CO
CO N
N
