

Astruc D. - Modern arene chemistry (2002)(en)
.pdf12.5 CpFeþ-Induced Triallylation of Toluene and Reactivity of the Triallyl Tripod Towards Transition Metals 413
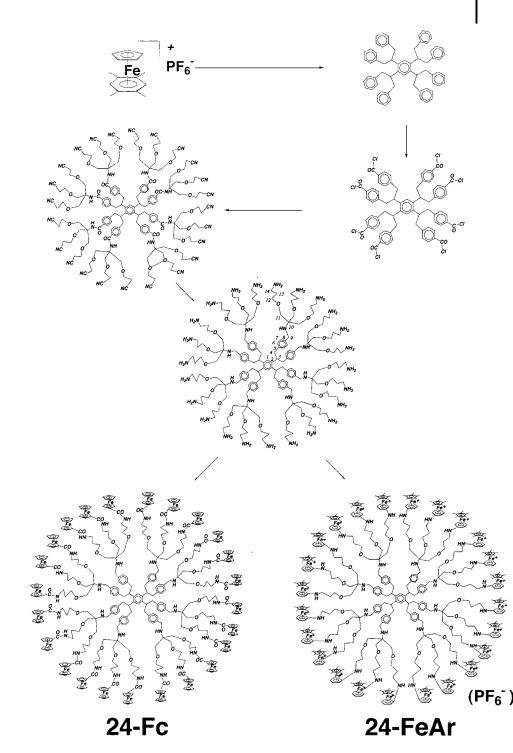
Scheme 13. Synthetic scheme for 24-CN and octa-ferrocenyl dendrimers.
decaisopropyland decaisopentyl cyclopentadienyl cobalt and rhodium complexes [56–58]) (Schemes 16 and 17).
12.5
CpFeB-Induced Triallylation of Toluene and Reactivity of the Triallyl Tripod Towards Transition Metals
In all of the above examples, the polybranching reaction of arene ligands was limited by the steric bulk. In the toluene and mesitylene ligands, however, the deprotonation–allylation re-

414 12 Activation of Simple Arenes by the CpFeþ Group
Scheme 14. Synthesis of a small dendrimer terminated by eight CpFeþ(h6-pentamethylbenzene) moieties starting from CpFeþ(h6- 1,2,4,5-tetramethylbenzene). The octanuclear compound obtained is almost insoluble in all solvents.
actions are no longer restricted by the neighborhood of other alkyl groups. All of the benzylic protons, i.e. three per benzylic carbon, can be replaced by methyl or allyl groups in the onepot iterative methylation or allylation reactions. Thus, the toluene complex can be triallylated and the resulting tripod can be desymmetrized by stoichiometric or catalytic reactions with transition metals, as shown in Scheme 18.
12.6
Nonaallylation of Mesitylene for the Synthesis of Dendritic Precursors of Large Metallodendrimers
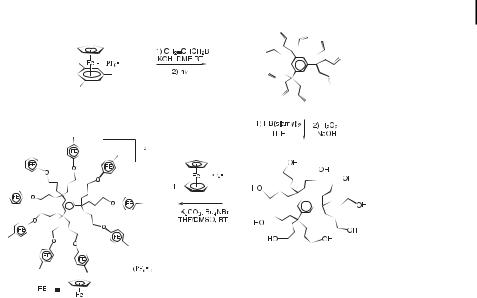
The mesitylene complex can be nonaallylated, the reaction proceeding smoothly at room temperature in the presence of excess KOH and allyl bromide (Scheme 19). The nonaallyl complex was photolyzed using visible light to remove the metal group CpFeþ, then hydroborated using 9-BBN, and the nonaborane was oxidized to the nonaol using H2O2/OH . Reaction of this nonaol with [FeCp(h6-p-MeC6H4F)][PF6] in the presence of K2CO3 in DMF yielded the nona-sandwich complex as a result of CpFeþ-induced nucleophilic substitution of the halogen by the alkoxy branch, and the metallodendrimer was subsequently purified by column chromatography (Scheme 19). The cyclic voltammogram of this complex shows that
12.6 Nonaallylation of Mesitylene for the Synthesis of Dendritic Precursors of Large Metallodendrimers 415
Scheme 15. Construction of dendrimers with 24 ferrocenyl or cationic iron-sandwich groups.

416 12 Activation of Simple Arenes by the CpFeþ Group
Scheme 16. Deca-alkylation, -allylation, or -ben- zylation of 1,2,3,4,5-pentamethylcobaltocenium or 1,2,3,4,5-pentamethylrhodocenium in a one-pot
reaction consisting of ten deprotonation–alkylation
sequences. Steric constraints inhibit further reaction, and the ten groups introduced are selforganized according to a single directionality (see Scheme 17).
Scheme 17. Interconversion between the clockwise and counterclockwise directionalities of the decaalkylated or decafunctionalized 18-electron cationic metallocenes (from the reactions in Scheme 11) as observed by 1H NMR. The coalescence temperature increases with increasing size of the introduced R group.
Scheme 18. One-pot CpFeþ-induced triallylation of toluene and reactions of transition metal complexes with the triallylated complex [59, 95].
12.6 Nonaallylation of Mesitylene for the Synthesis of Dendritic Precursors of Large Metallodendrimers 417
Scheme 19. One-pot, large-scale, high-yielding CpFeþ-induced nonaallylation of mesitylene and subsequent functionalization of the olefinic branches leading to redox-active nonametallic dendrimers.
the nine redox FeII centers are seemingly reduced at the same potential. At 30 C, this wave is reversible and the number of redox centers is found to be 8 G1 using the Bard– Anson formula [59, 60].
Alternatively, Michael reaction of the nonaol with acrylonitrile yields a nonanitrile, which can be reduced to the nonaamine. This nonaamine was allowed to react with chlorocarbonylmetallocenes and other chlorocarbonyl sandwich complexes to yield nonaamidometallocenes [61–63] and nonaamido-sandwich compounds [63] (Scheme 20). These metallodendrimers also give rise to only one cyclic voltammetry wave, the intensity of which corresponds to approximately nine electrons using the technique indicated above (the solvent was CH2Cl2 for the nonaamidoferrocene and MeCN for the polycationic dendrimers). Chemical reversibility was observed at room temperature, although some adsorption was noted.
The nonaamine was the starting point for the construction of dendrimers according to the iterative procedure first reported by Vo¨gtle, and then developed by the groups of Meijer [64] and Mu¨lhaupt [65]. Thus, reaction of the nonaamine with acrylonitrile gave an 18-nitrile dendrimer, which was reduced to the 18-amine dendrimer. The dendritic construction was continued in this way until the 144-nitrile dendrimer was reached. Purification of these polynitrile dendrimers was achieved at each generation by column chromatography [66]. Each polyamine dendrimer reacted with chlorocarbonylferrocene, but the 36and 72-amidoferrocene dendrimers obtained were found to be insoluble in all solvents tested. The last soluble amidoferrocene dendrimer of this series is the 18-amidoferrocene (Scheme 21). This dendrimer subsequently proved very e cient for anionic recognition [61, 62].

418 12 Activation of Simple Arenes by the CpFeþ Group
Scheme 20. Synthesis of nonaamidometallocenyl dendrimers using the core created by the CpFeþ-induced nonaallylation of mesitylene.

12.7 CpFeB-Induced Activation of Ethoxytoluene in the One-Pot Synthesis of a Phenol Dendron 419
Scheme 21. Syntheses of cationic nonaamido-sandwich complexes of iron and cobalt (18-electron forms) using the same core as that used in Scheme 20.
12.7
CpFeB-Induced Activation of Ethoxytoluene in the One-Pot Synthesis of a Phenol Dendron by Triple-Branching and Synthesis of Organometallic Dendrons
As the triple-branching reaction shown in Scheme 18 is very straightforward, we sought a more sophisticated version compatible with a functional group in the para position of the tripod in order to provide access to a functional dendron. Serendipitously, we found that KOH or tBuOK under very mild conditions easily cleaved the iron complexes of aromatic ethers. The activating CpFeþ group again induces this reaction, which is very general for a variety of aromatic ether complexes (Scheme 22) [67, 68].
Since this cleavage reaction is carried out with the same reagent and solvent as those used in the trialkylation reaction (ideally tBuOK in THF), we attempted to perform both reactions in a well-defined order (triallylation before ether cleavage) in a one-pot reaction. Indeed, this worked out well, and the Feþ complex of the phenol tripod was prepared in 60 % yield in this way. This complex could be photolyzed in the usual way using visible light, yielding the free phenol tripod. However, we also investigated the possibility of cleaving the arene ligand

420 12 Activation of Simple Arenes by the CpFeþ Group
Scheme 22. Heterolytic CaO cleavage reaction in aryl ether complexes by tBuOK or KOH, induced by the activating 12-electron fragment CpFeþ. This reaction is very useful and has been applied to the convenient onepot synthesis of the phenol-triallyl dendron (see Scheme 23).
in situ at the end of the phenol tripod construction. tBuOK is a reductant when it cannot perform other reactions. Since the two important reactions had been completed, it was envisaged that the third role of tBuOK, as a single-electron reductant, would come into play. This reasoning turned out to be correct. The cleavage of the arene intervenes rapidly at the 19-electron stage, because 19-electron complexes of this kind are not stable with a heteroatom located in an exocyclic position (most probably because the heteroatom coordinates to the metal of the labile 19-electron structure). After optimizing the reaction conditions, a 50 % yield of the free phenol dendron could be reproducibly obtained from the ethoxytoluene complex [50, 69], and this reaction is now regularly used in our laboratory to synthesize this very useful dendron as a starting material (Scheme 23).
Scheme 23. One-pot syntheses of the phenol-triallyl iron complex and metal-free dendron by variation of the experimental conditions. The iron complex can be demetalated by visible-light photolysis; the metal-free phenol-triallyl dendron can be obtained more rapidly direct from the p- ethoxytoluene-iron complex.
Functionalization of the three allyl chains of the phenol dendron could be achieved by hydrosilylation, as catalyzed by the Karsted catalyst [70]. Indeed, it is very interesting that there is no need to protect the phenol group before performing these reactions. For instance, catalyzed hydrosilylation using ferrocenyldimethylsilane gives a high yield of the triferro-

12.8 Convergent and Divergent Syntheses of Large Ferrocenyl Dendrimers with Good Redox Stabilities 421
cenyl dendron HO-p-C6H4C(CH2CH2CH2SiMe2Fc)3, which can easily be purified by column chromatography [70, 71].
12.8
Convergent and Divergent Syntheses of Large Ferrocenyl Dendrimers with Good Redox Stabilities
Protection of the phenol dendron using propionyl iodide gave the phenolate ester, which was hydroborated, and then oxidation of the triborane using H2O2/OH gave the triol. Reaction with Me3SiCl gave the tris-silyl derivative, treatment of which with NaI yielded the triiodo compound, and reaction of the latter with the triferrocenyl dendron provided the nonaferrocenyl dendron, which was deprotected using K2CO3 in DMF. The nonaferrocenyl dendron was allowed to react with hexakis(bromomethyl)benzene, which gave the 54-ferrocenyl dendrimer. This convergent synthesis proceeded cleanly and the 54-ferrocenyl dendrimer gave correct analytical data, although a mass spectrum could not be obtained (Scheme 24).
This approach is somewhat limited, however, since larger dendrons that one might like to synthesize in this way cannot be made because dehydrohalogenation becomes faster than nucleophilic substitution of the iodo by phenolate in the bulkier higher-generation dendrons. Although this problem might be overcome by modifying the iodo branch in such a way that there are no hydrogens in the b-positions, the condensation of higher dendrons onto a core would become tedious or impossible for steric reasons. This well-known inconvenience is intrinsic to the convergent dendritic synthesis. On the other hand, divergent syntheses are not marred by such a problem since additional generations and terminal groups are added at the periphery of the dendrimer. The limit is that indicated by De Gennes, i.e. the steric congestion encountered at the generation at which the peripheral branches can no longer be divided. Another obvious limitation arises if the molecular objects added onto the termini of the branches are large and interfere with one another. We have developed a divergent synthesis of polyallyl dendrimers, as indicated in Scheme 20, whereby each generation is created by a sequence of hydroboration, oxidation of the borane to the alcohol, formation of the mesylate, and reaction of the phenol dendron with the mesylate. This strategy has allowed us to synthesize dendrimers of generations 0, 1, 2, and 3 with 9 (G0), 27 (G1), 81 (G2), and 243 branches (G3), respectively (Scheme 25 and Chart 1).
The MALDI-TOF mass spectrum of the 27-allyl dendrimer shows only the molecular peak and traces of a side product. That of the 81-allyl dendrimer shows a dominant molecular peak, but also peaks due to significant amounts of side products resulting from incomplete branching. That of the 243-allyl dendrimer could not be obtained, possibly signifying that this dendrimer is polydisperse (correct 1H and 13C NMR spectra were nevertheless obtained, indicating that the ultimate reactions had proceeded to completion). This dendrimer proved to be soluble, indicating that this generation is not the last one that might be reached. Ferrocenylsilylation of all these polyallyl dendrimers was accomplished using ferrocenyldimethylsilane, as catalyzed by the Karsted catalyst [72] at room temperature or 40 C. The reactions were complete after a day, except in the case of the ferrocenylsilylation of the 243allyl dendrimer, which required a reaction time of two weeks indicating some degree of steric congestion (Schemes 26 and 27).
The 1H and 13C NMR spectra indicate the absence of regioisomers. Solubility in pentane

422 12 Activation of Simple Arenes by the CpFeþ Group
Scheme 24. Convergent strategy for the clean synthesis of a 54-ferrocenyl dendrimer. This scheme also illustrates the ability of such large ferrocenyl dendrimers to serve as molecular batteries. The deep-blue ferrocenium form can be synthesized
quantitatively, characterized, and reduced back to the orange ferrocenyl dendrimer. The overall cycle proceeds without any decomposition with all these silylferrocenyl dendrimers, including the 243ferrocenyl dendrimer.