

Astruc D. - Modern arene chemistry (2002)(en)
.pdf14.4 Other Reagents for the Oxidative Coupling Reaction 513
Fig. 3. The putative ortho-palladated benzoate intermediate in the dimerization of methyl benzoate.
14.4.6
Non-Metal Mediated Methods
Wul and co-workers observed that the oxidative dimerization of 147 can be accomplished extremely e ciently when it is melted in a sealed tube in the presence of air, to furnish the biaryl product 148, a precursor to the natural compound gossypol (149) (Scheme 35) [99]. The same reaction with iron(III) chloride gives low yields and a less clean product distribution. It is interesting to note that in the case of 3-phenyl-1-naphthol (150), the regioselectivity of the iron(III) chloride-mediated oxidation is completely di erent from that observed with O2 as the oxidant, with the para-para-coupled product 152 being favored. This air oxidation procedure is also applicable to phenanthrol units (e.g. 153 ! 154), giving similarly high yields.
Scheme 35. Oxygenand iron(III)-mediated dimerization of naphthol and phenanthrol derivatives.
Horseradish peroxidase (HRP) has also been used in an enzymatic process to create aryl– aryl bonds, especially for the synthesis of diiodotyrosine derivatives in a possibly biomimetic transformation [30, 100]. Here again, the regioselectivity of bond formation is an important issue and, depending on both the conditions and the substrate, CaC or CaO-coupling can occur. The e ect of substituents ortho to the alcohol group has been studied by Sih and co-


14.5 Phase-Supported Oxidants 515
It is noteworthy that naphthalene itself does not react e ciently (6 %) under these conditions and that 1-naphthol is too reactive, even at 78 C. Steric hindrance is also an important factor that tends to lower the yields and the regioselectivity. The removal of an electron from the aromatic ring by NOþ triggers the coupling reaction. Oxygen is also involved in the proposed mechanism (Scheme 37).
Scheme 37. Proposed mechanistic course of the NOþ-mediated dimerization of 1-methylnaphthalene.
14.5
Phase-Supported Oxidants
14.5.1
Reagents Supported on Inorganic Materials
A dispersion of copper sulfate on alumina has been studied by Sakamoto and co-workers for the oxidative dimerization of 2-naphthol derivatives [102, 103]. This heterogeneous reagent leads to the formation of binaphthols in very high yields (Table 32). These results show that the particular heterogeneous conditions indicated are favorable for the coupling of 2- naphthols. In related work, Lakshmi and co-workers have shown that Cu2þ-exchanged montmorillonite is an e cient supported copper reagent for the oxidative dimerization of naphthols (Table 32) [104]. Compared to all other methods of binaphthol formation, these supported phases certainly o er practical advantages. The products formed are easily isolated from the solid phase, which can be reactivated and reused.
One-electron oxidation of the naphthol substrate by copper(II) sulfate and coupling of the derived cation radicals seem to be the central steps of this reaction. The presence of dioxygen in the mixture may be important for the reactivation of the copper catalyst (oxidation of Cu(I) to Cu(II)).
Iron(III) chloride has also been dispersed onto alumina and montmorillonite to perform the same kind of reaction [104, 105]. In a study of these reagents, Li and co-workers showed that similar yields could be obtained in very short reaction times (Table 33).
Zeolites (crystalline aluminosilicates) supporting oxidizing metals (Fe or Cu) have been introduced by Garcia and co-workers [106]. In particular, they showed that FeMCM-41 affords binaphthol from 2-naphthol as virtually the only product. Unfortunately, this compound is trapped in the zeolite pores and the isolated yields are lower than expected.
14.5.2
Polymer-Supported Hypervalent Iodine Reagents
Ley and co-workers studied the applicability of immobilized phenyliodine(III) bis(acetate) as an oxidative coupling reagent in an e ort to develop orchestrated multistep syntheses using

51614 Oxidative Aryl-Coupling Reactions in Synthesis
Tab. 32. Alumina and montmorillonite-supported copper(II)-mediated oxidative dimerization of 2-naphthol derivatives.
Substrate |
Time (h) |
|
Yield (%) |
|
|
|
|
|
|
|
|
|
Al2O3 |
montmorillonite |
|
|
|
|
|
68a |
8 |
97 |
95 |
|
68b |
2 |
98 |
91 |
|
68m |
4 |
98 |
98 |
|
68n |
15 |
93 |
40 |
|
68i |
8 |
97 |
91 |
|
68k |
18 |
|
41a(94)b |
– |
68f |
8 |
Trace(20)c |
– |
|
68o |
8 |
|
d |
– |
|
|
|||
68d |
8 |
|
e |
– |
|
|
|||
|
|
|
|
|
a Hexyl acetate used as solvent;
b at 160 C for 8 h in hexyl acetate, CuSO4/68k ¼ 2; c At 160 C, CuSO4/68f ¼ 2;
d No reaction;
e Complex mixture of products obtained.
polymer-supported species [107–109]. They successfully applied this strategy in the synthesis of the alkaloids (G)-oxomaritidine (159) and (G)-epimaritine (160) (Scheme 38). The use of polymer-supported PIFA led to similar yields as those achieved with polymer-supported PIDA in this oxidative coupling reaction. This interesting extension of hypervalent iodine chemistry furnished the coupled products in yields comparable to those achieved using the
Scheme 38. Polymer-supported hypervalent iodine-mediated oxidative cyclization of a maritidine precursor.

14.6 Control of Atropisomerism 517
Tab. 33. Supported iron(III)-mediated oxidative dimerization of 2-naphthol (68a) and its 6-bromo analogue (68b).
Catalyst (equiv.) |
Substrate |
Time (min) |
Yield (%) |
|||
|
|
|
|
|||
FeCl3/Al2O3 (0.2) |
68a |
20 |
90 |
|||
FeCl3 |
/Al2O3 |
(2.0) |
68a |
10 |
94a |
|
FeCl3 |
/Al2O3 |
(0.2) |
68b |
10 |
91 |
|
Fe3þ/mont. (0.2) |
68a |
180 |
93 |
|||
Fe3þ/mont. (0.2) |
68b |
50 |
98 |
|||
FeCl3 |
/SiO2 |
(0.2) |
68a |
150 |
98 |
|
FeCl3 |
/SiO2 |
(0.2) |
68b |
180 |
98 |
|
a Carried out in toluene at 60 C under nitrogen.
homogeneous version of the reaction (see Section 14.3), with the environmental advantage of a recoverable phase-supported reagent.
14.6
Control of Atropisomerism
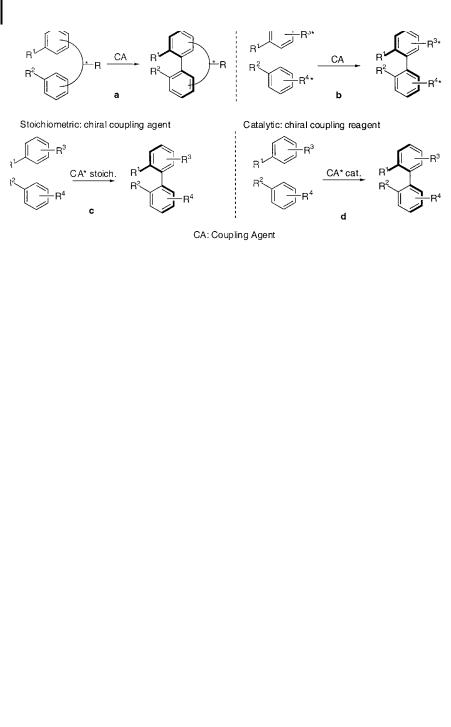
In some instances of biaryl formation, ortho,ortho0 substituents are su ciently sterically encumbering to restrict rotation about the AraAr linkage. These systems display axial chirality, and the process of atropisomerism through AraAr bond rotation becomes important [110, 111]. Controlling the atrop-selectivity of an oxidative coupling reaction is a key criterion for success in many natural products synthesis endeavors. Such control in biosynthesis is implied, of course, by the existence of single diastereomers of the many biaryl-containing natural principles, and consequently in vitro mimicry of this stereochemical feature is a central element of many biomimetic approaches to natural product targets. To date, two distinct ways of achieving such control have been explored. On the one hand, stereochemical information can be transferred from resident chirality in the precursor. The existing stereogenic center(s) can be incorporated within a backbone element of the structure or can be attached via a removable tether to the molecular core (Scheme 39a). Other related approaches introduce a stereochemical bias by attaching a chiral auxiliary directly to one (or both) of the aryl rings (Scheme 39b). A conceptually distinct strategy for fixing atropisomer stereochemistry upon AraAr bond formation involves the use of a chiral oxidation reagent (Scheme 39c). More interestingly, a catalytic version of these asymmetric reactions can also be envisaged (Scheme 39d). Many publications have appeared recently detailing progress on the first cases (chiral information built into the starting material). In addition, very interesting work relating to the second category (chiral oxidants) and, most recently, catalytic enantioselective couplings have been described.


520 14 Oxidative Aryl-Coupling Reactions in Synthesis
Fig. 5. Examples of stegane-type lignans.
[122, 123], schizandrin (169), and gomisin-A (170) [124] contain biaryl structures as part of a dibenzocyclooctadiene unit (Figure 5), making them compelling targets for biaryl oxidative coupling methodologies.
The control of atrop-selectivity upon oxidative coupling has been more challenging in these cases due to the fact that the energy barrier between the two atropisomers is rather low. The natural molecules themselves are sometimes actually present in the two forms (e.g. (þ)-schizandrin and (þ)-isoschizandrin). In some instances, the non-natural atropisomer is found to be the major coupling product under kinetic control of the reaction, even if the precursor bears the natural stereogenic centers on the butyrolactone moiety [111]. For a para-substituted phenol precursor such as 171, the mechanism of oxidative coupling involves the formation of a spirodienone intermediate 172. The subsequent rearrangement of this species to the biaryl products 173 and 174 then occurs with migration of the indicated CaC bond in one of two directions (a or b), leading to a mixture of atropisomers usually favoring the non-natural configuration shown in 174 (Scheme 42a) [125]. Buckleton and co-workers have proposed another mechanism for non-phenolic substrates such as 175. In this case, they hypothesized that either one ‘‘two-electron’’ oxidation (175 ! 178) or two ‘‘one-electron’’ steps (175 ! 176 ! 176a) can lead to the dication 177, which then forms the biaryl 179 by proton loss (Scheme 42b). Buckleton posits that the interactions between bridgehead hydrogens (away from each other in intermediate 177 (Scheme 42)) favor the isostegane-like series [37].
Ward, Pelter, and co-workers have documented the e cacy of in situ generated ruthenium tetrakis(trifluoroacetate) (following the work of Robin and Landais) [126, 127] as a useful reagent for the synthesis of a large number of stegane-like molecules [123]. In the transfused butyrolactone series 180, single diastereomers 181 are usually obtained (Scheme 43a). If cis-fused butyrolactones 182 are used as precursors, the biaryl products are formed as a mixture of atropisomers 183 and 184 (Scheme 43b).
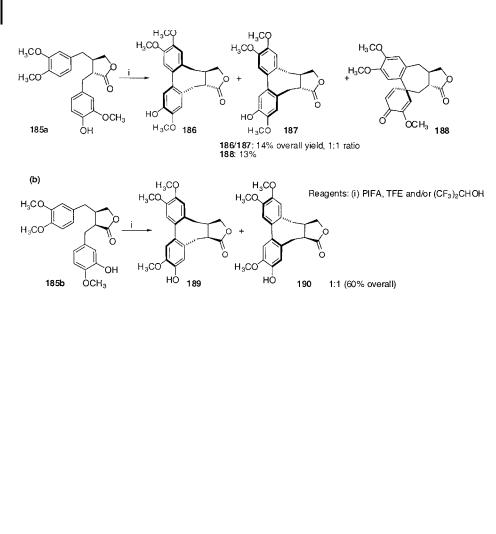
In parallel studies to the ruthenium-based work, Ward and co-workers have also shown that PIFA (Scheme 44) [122, 128] and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) (Scheme 45) [122, 129, 130] are e ective reagents for coupling the phenolic precursors 185a,b. Yields are generally lower than in the fully methylated case (spirodienone products 188 can be formed when a hydroxyl group is present at the para position of the 2-benzyl group of 185a) and unbiased mixtures of atropisomers 186/187 and 189/190 are obtained.

522 14 Oxidative Aryl-Coupling Reactions in Synthesis
Scheme 44. PIFA-mediated oxidative cyclization of transand cis-butyrolactone stegane precursors.
Scheme 45. DDQ-mediated oxidative cyclization of transand cis-butyrolactone stegane precursors.
The DDQ-mediated transformations of 180a–c are oxidative couplings that may actually proceed through nucleophilic capture of a benzylic cation (or an equivalent thereof ) 193 by the adjacent aryl ring. Initial one-electron aryl oxidation (180 ! 191) followed by proton loss (191 ! 192) and subsequent one-electron removal may precede benzyl cation 193 formation, as shown in Scheme 46 [129].
Further examples of oxidative arylic coupling within this family of lignan precursors include the preparation of (þ)-schizandrin (169) and (þ)-gomisin A (170) (Figure 5), as published by Tanaka and co-workers. In this instance, the key AraAr coupling step with 194 was realized using either ruthenium tetrakis(trifluoroacetate) or iron perchlorate (Scheme 47) [124, 131–133]. As pointed out by the authors, this result was noteworthy as this simple