

Astruc D. - Modern arene chemistry (2002)(en)
.pdf9.3 {TpRe(CO)(L)} 323
Fig. 7. Formation of an h2-o-quinone methide complex from an aldol condensation of crotonaldehyde and the h2-phenol complex 85.
93 and 96, respectively. Although isolated examples of C2 addition have been observed for complexes of 2-methyl anisole [20] and N-ethyl aniline [27], C2 alkylation of [Os]-arenes is primarily a reactivity pattern unique to phenols.
9.2.8.3 o-Quinone Methide Complexes
Another example of C2 alkylation is the generation of o-quinone methide complexes from [Os]-phenol and crotonaldehyde. When these reagents are combined in the presence of BF3 OEt2, addition occurs at C4 (Table 14). In the absence of a catalyst, an aldol condensation occurs at C2 to generate the o-quinone methide complex 97 in 85 % yield (Figure 7) [29]. This reaction appears to be general for aldehydes and h2-phenol complexes, even when the phenol is not substituted at C4.
9.3
{TpRe(CO)(L)}
9.3.1
Introduction
The [Os] fragment is a powerful tool for the dearomatization and activation of aromatic molecules. For more than a decade, however, alternative dearomatizing agents have been sought because of inherent limitations associated with the [Os] system. The metal fragment is achiral and therefore cannot discriminate between enantiofaces upon binding. Unfortunately, changes to the ligand set about the metal completely nullify its ability to coordinate aromatic molecules [31]. Consequently, the achiral nature of the metal center as well as the electronic and steric properties of the system cannot be adjusted. In addition, the dicationic charge of the [Os] complexes limits the solvent environments and purification techniques that may be utilized.
Investigations of cationic rhenium(I) complexes with varying ligand sets showed that the inclusion of a single p-acid in the ligand set was necessary in order to approximate the electronic characteristics of the [Os] fragment [32]. The rhenium(I) fragment facfRe(dien)(PPh3)(CO)gþ (dien ¼ diethylenetriamine) was successfully bound to furan in an h2-fashion. However, it could not be made to bind arenes, with the large PPh3 ligand in the coordination sphere presumably causing a significant steric hindrance.
Ultimately, it was the Tp ligand, a strong s-donor (L ¼ PMe3), and the p-acid (CO) that proved to possess the right combination of steric and electronic properties to facilitate the h2- binding of a variety of aromatic molecules to an Re(I) center [33, 34]. Further studies led to

3249 Osmiumand Rhenium-Mediated Dearomatization Reactions with Arenes
a series of Re(I) dearomatization fragments having the general formula fTpRe(CO)(L)g (L ¼ tert-butyl isonitrile (tBuNC), PMe3, pyridine (py), N,N-dimethylaminopyridine (DMAP), N-methylimidazole (MeIm), or NH3). These fragments not only allow the adjustment of the steric and electronic properties through variation of L, but also have the potential for enantiofacial discrimination of bound prochiral aromatic molecules.
9.3.2
Preparation of h2-Arene Complexes
Metal fragments of the form fTpRe(CO)(L)g, in which L ¼ MeIm or NH3, have the ability to bind substituted benzenes in an h2-fashion. The fragment having L ¼ MeIm has seen more
application because of di culties associated with the synthesis and isolation of aromatic
complexes having L ¼ NH3. Anisoles, phenols, and naphthalenes all form thermally stable h2-complexes when stirred in excess (10–25 equiv.) with TpRe(CO)(MeIm)(h2-benzene) in
THF. The complexes are typically isolated by precipitation from hexanes. The benzene
complex can be prepared by direct reduction of the Re(III) precursor, TpRe(MeIm)(Br)2, with Nao in benzene under one atmosphere of CO [35].
Naphthalene forms stable h2 complexes with fTpRe(CO)(L)g when L ¼ tBuNC, PMe3, py, DMAP, or MeIm. In toluene and an excess of naphthalene, direct reduction of TpRe(L)Br2 (L ¼ py or DMAP) with Na0 under CO atmosphere generates the py or DMAP naphthalene complexes in @40 % yield [36, 37]. Generation of the Re(I) PMe3 and tBuNC naphthalene systems is not as straightforward. They are synthesized by oxidation of the corresponding Re(I) alkene complexes to Re(II), liberation of the alkene by heating, and reduction of the Re(II) species in the presence of naphthalene [36].
9.3.3
Quadrant Analysis
Upon complexation to the fTpRe(CO)(L)g fragment, a coordinated double bond becomes oriented orthogonally to the ReaCO bond and approximately parallel to the ReaL bond. Such an orientation is thought to maximize overlap between the d orbitals of the metal and the p orbitals of the CO and the h2-bound species [34]. This arrangement limits the number of coordination diastereomers to two, each of which exists as one dominant rotamer. Typically, the bound arene projects into quadrants a and d (Figure 8) in order to minimize steric interactions with the scorpionate ligand [34]. The proton labeled HA on the pyrazolyl ring trans to the CO is directed forward from the quadrant plane, as indicated in Figure 8, and inhibits the placement of any substituents in quadrants b or c. A study of the binding selectivities of substituted alkenes and ketones with L ¼ MeIm suggested that quadrant c is the most sterically congested and that quadrant d is the least sterically congested [38, 39]. Thus, only one rotamer is typically observed for each coordination diastereomer (vide infra).
9.3.4
Naphthalene
In solution, naphthalene complexes of the form TpRe(CO)(L)(h2-naphthalene) exist as mixtures of diastereomers A and B (Table 15). Only when L ¼ PMe3 (entry 1) does the unbound ring of the naphthalene show a thermodynamic preference for quadrant a (98A). For all

9.3 {TpRe(CO)(L)} 325
Fig. 8. Quadrant analysis of fTpRe(CO)(L)g fragments.
other L’s, the preference is for the unbound ring to lie in quadrant d (98B) (vide supra). When exposed to a solution of HOTf in acetonitrile followed by MMTP, complex 98 undergoes tandem electrophilic/nucleophilic addition reactions with a high degree of regiocontrol. Oxidative decomplexation with AgOTf at room temperature liberates the dihydronaphthalenes
Tab. 15. Regioisomeric ratios and yields of dihydronaphthalenes.
Entry |
L |
T ( C) |
98A:98B |
Method A* |
|
Method B** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
99:100 |
Yield |
99:100 |
Yield |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
PMe3 |
0 |
– |
– |
– |
13:1 |
29 % |
|
|
|
20 |
10:1 |
12:1 |
52 % |
12:1 |
24 % |
|
|
|
8 |
|
|
|
|
|
|
2 |
Pyridine |
< 40 |
– |
1:15 |
63 % |
– |
– |
|
|
|
0 |
– |
– |
– |
1:7 |
80 % |
|
|
|
: 20 |
1:3 |
1:8 |
89 % |
1:7 |
66 % |
|
|
|
|
|
|
|
|
|
|
3 |
DMAP |
40 |
– |
1:25 |
19 % |
– |
– |
|
|
|
20 |
1:1.5 |
1:10 |
50 % |
– |
– |
|
4 |
|
|
|
|
|
|
|
|
Melm |
0 |
– |
– |
– |
1:25 |
55 % |
||
|
|
20 |
1:5 |
1:23 |
83 % |
1:16 |
63 % |
|
|
|
|
|
|
@25 % |
|
|
|
5 |
NH3 |
40 |
– |
1:7 |
– |
– |
||
|
|
20 |
1:4 |
– |
– |
– |
– |
|
|
|
|
|
|
|
|
|
|
* ¼ Reaction sequence performed in inert atmosphere. ** ¼ Reaction sequence performed on benchtop, with @500 mg of metal complex, and blanketed under dinitrogen.

3269 Osmiumand Rhenium-Mediated Dearomatization Reactions with Arenes
99 and 100. When L ¼ PMe3 (entry 1), the reaction favors the 1,4-addition product (99), a result similar to that observed with [Os]-bound naphthalene. Complexes that do not have L ¼ PMe3 (entries 2–5) generate the 1,2-addition product (100) with di erent degrees of selectivity [37]. The regioselectivity of the addition is highest for L ¼ MeIm (25:1), but the overall yield is highest for L ¼ py (89 %). Notably, these reactions do not require a glovebox and can be performed with @500 mg of metal complex (Method B). A high degree of stereocontrol has also been demonstrated in these reactions, with both the electrophile and the nucleophile adding anti to the face involved in metal coordination. Furthermore, despite the initial presence of two coordination diastereomers, a single coordination diastereomer can be isolated from each reaction prior to oxidation of the metal center. This result indicates a potential for the synthesis of enantioenriched dihydronaphthalenes through the use of resolved metal complexes [39].
9.3.5
Cycloadditions
Another early success of the fTpRe(CO)(MeIm)g fragment was the promotion of Diels– Alder cycloaddition reactions with benzene and anisole. Complex 101 [TpRe(CO)(MeIm)(h2- benzene)] undergoes an endo-selective Diels–Alder reaction with N-methylmaleimide to a ord the bound bicyclo[2.2.2]octadiene complex 102 in 65 % yield (Scheme 12) [40]. Oxidation of 102 yields the bicyclo[2.2.2]octadiene 103 and/or the bicyclo[2.2.2]octenone 104 depending upon the choice of oxidation conditions.
Scheme 12. The fTpRe(CO)(MeIm)g-promoted Diels–Alder reaction of benzene.
The more activated TpRe(CO)(MeIm)(h2-anisole) complex has demonstrated a slightly broader range of reactivity by undergoing cycloadditions with both N-methylmaleimide (Scheme 13) and dimethylacetylene dicarboxylate (DMAD) (Scheme 14). Analysis of these reactions is complicated by the fact that the initial anisole complex exists as a 3:1 mixture of

9.3 {TpRe(CO)(L)} 327
Scheme 13. A fTpRe(CO)(MeIm)g-promoted Diels–Alder reaction of anisole and N-methylmaleimide.
coordination diastereomers (Scheme 13). The major diastereomer 105A has the methoxy group of the anisole ring directed toward the MeIm ligand, while the minor diastereomer 105B has the methoxy group directed away from the MeIm ligand. The N-methylmaleimide cycloadducts 106A and 106B are formed in a diastereomeric ratio of 1:1. They can be sepa-
Scheme 14. A fTpRe(CO)(MeIm)g-promoted Diels–Alder reaction of anisole and DMAD.

3289 Osmiumand Rhenium-Mediated Dearomatization Reactions with Arenes
rated by chromatography and are far less stable to cycloreversion than the corresponding benzene cycloadduct (t1=2 ¼ 1 h for 106A; t1=2 ¼ 0.5 h for 106B). Interestingly, initial studies indicate that the decomplexation of these two adducts yields two di erent oxygen insertion products (107 and 108). Oxidation of complex 106A leads to oxygen insertion into the exoalkene (107), while oxidation of complex 106B leads to oxygen insertion into the endo-alkene (108).
Complex 105 cyclizes with DMAD to give the diastereomeric bound bicyclo[2.2.2]octatrienes 109A and 109B in a 1:1 ratio (Scheme 14). Oxidation of these complexes liberates the corresponding triene 110 as well as the disubstituted anisole 111, presumably generated with acetylene from the cycloreversion of 110.
9.4
Concluding Remarks
While more than a decade has passed since the first reports of reactions with h2-bound aromatic molecules, this field is still in its infancy compared to the more established field of h6- arene chemistry. Nevertheless, the general strategy of using a transition metal to render a conjugated p-system more electron-rich is now firmly established. The next phase of development will be marked by a greater focus on practical issues such as less expensive p-basic metal systems, e cient control of absolute stereochemistry, and standardization of procedures that allow for easier reagent handling.
Acknowledgement
Thanks are due to the many fine graduate, postgraduate, and undergraduate students whose labors have made this account possible.
References
1 |
J. P. Collman, L. S. Hegedus, J. R. |
7 |
W. H. Miles, P. M. Smiley, H. R. |
|
Norton, R. G. Finke, Principles and |
|
Brinkman, J. Chem. Soc., Chem. Commun. |
|
Applications of Organotransition metal |
|
1989, 659. |
|
Chemistry, University Science Books, Mill |
8 |
Jung-Nyoung Heo, Org. Lett. 2002, 2, |
|
Valley, CA, 1987, p. 409–415. |
|
2987. |
2 |
M. F. Semmelhack, in Comprehensive |
9 |
A. J. Pearson, J. G. Park, J. Org. Chem. |
|
Organic Synthesis, Vol. 4 (Eds.: B. M. |
|
1992, 57, 1744. |
|
Trost, I. Fleming, M. F. Semmelhack), |
10 |
D. Astruc, Tetrahedron 1983, 39, 4027. |
|
Pergamon Press, Oxford, 1990, p. 517. |
11 |
P. A. Lay, R. H. Magnuson, H. Taube, |
3 |
A. R. Pape, K. P. Kaliappan, E. P. |
|
Inorg. Synth. 1986, 24, 269. |
|
Kundig, Chem. Rev. 2000, 100, 2917. |
12 |
W. D. Harman, H. Taube, J. Am. Chem. |
4 |
A. J. Pearson, M. C. Milletti, P. Y. Zhu, |
|
Soc. 1988, 110, 7906. |
|
J. Chem. Soc., Chem. Commun. 1995, |
13 |
W. D. Harman, H. Taube, J. Am. Chem. |
|
1309. |
|
Soc. 1989, 112, 2261. |
5 |
A. J. Pearson, P. Y. Zhu, J. Am. Chem. |
14 |
D. W. Rogers, F. J. McClafferty, J. Org. |
|
Soc. 1993, 115, 10376. |
|
Chem. 2001, 66, 1157. |
6 |
A. J. Pearson, P. R. Bruhn, J. Org. Chem. |
15 |
F. H. Ding, W. D. Harman, unpublished |
|
1991, 56, 7092. |
|
results (2002). |
|
|
|
References |
329 |
|
|
|
|
|
16 |
M. D. Winemiller, M. E. Kopach, W. D. |
29 |
M. E. Kopach, W. D. Harman, J. Am. |
|
|
Harman, J. Am. Chem. Soc. 1997, 119, |
|
Chem. Soc. 1994, 116, 6581. |
|
|
2096. |
30 |
M. E. Kopach, L. P. Kelsh, K. Stork, W. |
|
17 |
M. D. Winemiller, W. D. Harman, J. |
|
D. Harman, J. Am. Chem. Soc. 1993, 115, |
|
|
Am. Chem. Soc. 1998, 120, 7835. |
|
5322. |
|
18 |
M. D. Winemiller, J. Org. Chem. 2000, |
31 |
J. Barrera, S. D. Orth, W. D. Harman, J. |
|
|
65, 1249. |
|
Am. Chem. Soc. 1992, 114, 7316. |
|
19 |
W. D. Harman, Chem. Rev. 1997, 97, 1953. |
32 |
B. C. Brooks, R. M. Chin, W. D. |
|
20 |
S. P. Kolis, M. E. Kopach, R. Liu, W. D. |
|
Harman, Organometallics 1998, 17, 4716. |
|
|
Harman, J. Org. Chem. 1997, 62, 130. |
33 |
T. B. Gunnoe, M. Sabat, W. D. Harman, |
|
21 |
M. E. Kopach, S. P. Kolis, R. Liu, J. W. |
|
J. Am. Chem. Soc. 1999, 121, 6499. |
|
|
Robertson, M. D. Chordia, W. D. |
34 |
T. B. Gunnoe, M. Sabat, W. D. Harman, |
|
|
Harman, J. Am. Chem. Soc. 1998, 120, 6199. |
|
Organometallics 2000, 19, 728. |
|
22 |
M. D. Chordia, W. D. Harman, J. Am. |
35 |
S. H. Meiere, B. C. Brooks, T. B. |
|
|
Chem. Soc. 2000, 122, 2725. |
|
Gunnoe, M. Sabat, W. D. Harman, |
|
23 |
M. D. Chordia, W. D. Harman, J. Am. |
|
Organometallics 2001, 20, 1038. |
|
|
Chem. Soc. 1998, 120, 5637. |
36 |
S. H. Meiere, B. C. Brooks, T. B. |
|
24 |
M. E. Kopach, W. D. Harman, J. Org. |
|
Gunnoe, E. H. Carrig, M. Sabat, W. D. |
|
|
Chem. 1994, 59, 6506. |
|
Harman, Organometallics 2001, 20, 3661. |
|
25 |
S. P. Kolis, M. E. Kopach, R. Liu, W. D. |
37 |
M. T. Valahovic, M. Sabat, W. D. |
|
|
Harman, J. Am. Chem. Soc. 1998, 120, |
|
Harman, J. Am. Chem. Soc. 2001, M. T. |
|
|
6205. |
|
Valahovic, T. B. Gunnoe, M. Sabat, |
|
26 |
S. P. Kolis, M. D. Chordia, R. Liu, |
|
W. D. Harman, JACS 2002, 124, 3309. |
|
|
M. E. Kopach, W. D. Harman, J. Am. |
38 |
S. H. Meiere, W. D. Harman, |
|
|
Chem. Soc. 1998, 120, 2218. |
|
Organometellics 2001, 20, 3876. |
|
27 |
S. P. Kolis, J. Gonzalez, L. M. Bright, |
39 |
S. H. Meiere, W. D. Harman, |
|
|
W. D. Harman, Organometallics 1996, 15, |
|
unpublished results (2001). |
|
|
245. |
40 |
M. D. Chordia, P. L. Smith, S. H. |
|
28 |
J. Gonzalez, M. Sabat, W. D. Harman, J. |
|
Meiere, M. Sabat, W. D. Harman, J. Am. |
|
|
Am. Chem. Soc. 1993, 115, 8857. |
|
Chem. Soc. 2001, 123, 10756. |
|

330
10
The Directed ortho Metalation Reaction – A Point of Departure for New Synthetic Aromatic Chemistry
Christian G. Hartung and Victor Snieckus
Abstract
After a brief allusion to the current status of synthetic aromatic chemistry, a tour based on the Directed ortho Metalation (DoM) strategy is provided with selections from recent literature which highlight a) the use of DoM for the regiospecific preparation of polysubstituted aromatics and heteroaromatics; b) the valuable symbiosis of DoM with transition metal catalyzed AraAr cross-coupling protocols; c) the extension of DoM to the Directed remote Metalation (DreM) concept in biaryls, which provides methodologies for the construction of condensed aromatics (fluorenones, phenanthrols) and heterocyclics (thioxanthones, xanthones, acridones, dibenzphosphorinones, dibenzthiepinone dioxides, dibenzoxepinones, dibenzazepinones, dibenzphosphinones); and d) the evolving links of DoM to other modern methodologies with a note on the DoM–Grubbs ring-closing metathesis connection. The presentation is solely methodologically rather than total synthesis oriented; dependable directed metalation groups (DMGs ¼ CONR2, OCONR2, SONR2, NHBoc), new and renewed, are emphasized; p-excessive (furan, thiophene, indole) and p-deficient (pyridine) heterocycle HetDoM chemistry is delineated. While definitely not comprehensive, this account attempts to give a flavor of current anionic aromatic chemistry in synthesis.
10.1
Introduction
‘‘The most common reaction of aromatic compounds is electrophilic aromatic substitution. Many di erent substituents can be introduced . . . Starting from only a few simple materials, we can prepare many thousands of substituted aromatic compounds.’’ J. McMurry, Organic Chemistry, 5th Ed., Brooks/Cole, Pacific Grove, CA, 2000, p. 592.
The study of the structure, synthesis, and reactivity of aromatic compounds has been one of the cornerstones of the teaching of organic chemistry. An account of the historical and sometimes disputed dream of Kekule´ [1] is followed by the beautiful logic of electrophilic aromatic substitution rules [2], which allows students to predict syntheses of sparsely substituted aromatics. Aside from a brief diversion into reactions of halonitrobenzenes with nucleophiles [3], this topic constitutes a large chapter in our 1st year organic education but by the time we teach upper year organic majors and graduate students, aromatic chemistry
Modern Arene Chemistry. Edited by Didier Astruc
Copyright 8 2002 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim ISBN: 3-527-30489-4

10.2 The DoM Reaction as a Methodological Tool 331
receives the label ‘‘classical’’, a certainty for further disregard and/or sparse coverage [4]. Although the preparation and simple reactions of PhMgX or PhLi are taught, aromatic carbanionic chemistry receives no mention in undergraduate texts; similarly, in advanced synthesis courses, with some exceptions [5], students are not familiarized with this subject.
Invariably, the first problem the fresh M.Sc. or Ph.D. student faces upon joining a pharmaceutical company is the construction of a complex, densely substituted aromatic or, even more commonly, heteroaromatic molecule as a starting material. The response is, let’s buy it [6]. Since, also invariably, this fails, the SciFinderTM or equivalent data base produces (hopefully) a number of hits based on familiar tactics which include (Scheme 1): Eþ substitution [2], Nu substitution (including SRN1 processes) [3], cycloaddition (usually Diels– Alder) [7], and de novo ring construction [8], which are familiar but not always fully appreciated. While also not accepted pedagogically, e orts by numerous groups over the past 25 years have added the Directed ortho Metalation (DoM) reaction to the armamentarium of the synthetic chemist as a general, regioselective, and e ective strategy for the rational construction of polysubstituted aromatic and heteroaromatic substances [9]. While examination of alternative routes is undeniable, the approach to the construction of a key aromatic starting material or intermediate by DoM is, with greater frequency, used for small-scale syntheses by the academic and medicinal chemist and is also increasingly accepted for multi-kg and higher scale process routes to drug candidates and commercial pharmaceuticals and agrochemicals.
Scheme 1. Synthetic approaches to substituted aromatics.
33210 The Directed ortho Metalation Reaction -- A Point of Departure for New Synthetic Aromatic Chemistry
Aims of this Account
This account aims to document, both conceptually and technically, the impact of DoM on the traditional practices of synthetic aromatic chemistry, and is partitioned into the following arbitrary modes from mainly recent research of various laboratories, including our own:
. DoM as a methodological tool for the synthesis of polysubstituted aromatics;
. Heteroaromatic Directed ortho Metalation (HetDoM) in synthetic methodology;
. DoM–transition metal catalyzed aryl–aryl cross coupling symbiosis;
. Directed remote Metalation (DreM) in aromatics and heteroaromatics;
. Evolving DoM connections to modern synthetic reactions (Sonogashira, Ullmann,
Buchwald–Hartwig, Grubbs metathesis).
10.2
The DoM Reaction as a Methodological Tool
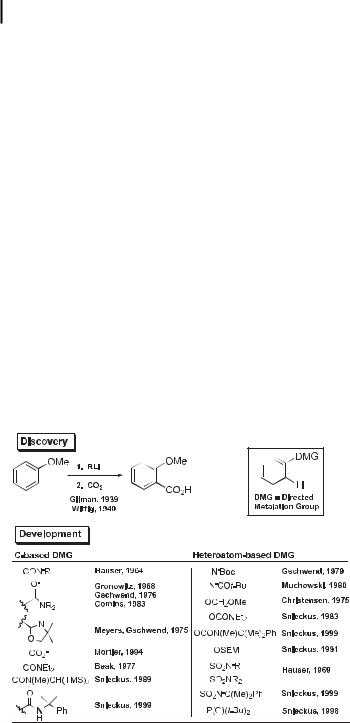
The concurrent and independent discovery of DoM by Wittig and Gilman over sixty years ago set the stage for the systematic work of Hauser, which was pursued in a number of laboratories in the late 1970s and has led to the current status of Directed Metalation Groups (DMGs), which allow the regioselective ortho introduction of a host of electrophiles (Scheme 2) [9a–c].
While the mechanism of DoM is an active and controversial field of study [10], a number of synthetic principles have emerged and are commonly practiced (Scheme 3). The con-
Scheme 2. Directed ortho Metalation: discovery and development.