

12. О неидеaльных рaстворaх. Модель с рaсслaивaнием.
12.1. Формальное описание неидеальных растворов. Избыточные функции и коэффициенты активности. Для формального описания неидеальных растворов применяют два метода. В первом методе вводится избыточная свободная энтальпия смешения Физб в соответствии с уравнением (9.1). Aналогичным образом опреде-ляется также избыточная энтальпия смешения—Низб–=–––T2[(Физб/Т)/T]p‚—избыточный объем смешения Vизб—=—(Физб/р)Т , избыточная энтрoпия смешения Sизб–=–––(Физб/Т)р и т.д. Величины Vизб и Qсмеш = – Низб называют также объемным и тепловым эффектами смешения. (Заметим‚ что в литературе теплота смешения иногда неясно определяется как “выделяемое или поглощаемое тепло”‚ а знак ее чаще выбирают в соответствии с указанной формулой‚ противоречащей общему правилу обозначений‚ которого желательно придерживаться во избежание путаницы: Q — теплота‚ получаемая системой‚ A — работа‚ совершаемая системой против внешних сил; конечно‚ и Q‚ и A могут быть положительными или отрицательными.)
При втором методе сохраняют для химических потенциалов формулы вида (8.11) или (9.2)‚ но заменяют концентрации хj активностями аj ‚ которые‚ таким образом‚ определены как
aj = exp [(j – jЧ ) / kT] (12.1)
или
aj = exp [(j – j0 ) / kT]. (12.2)
Формула (12.1) определяет активности однозначным образом. При таком определении они совпадают с хjв чистых и почти чистых компонентах. Определение (12.2) оставляет произвол. При этом определении выбираютj0 иаj для растворенных веществ так‚ чтобы в предельно разбавленном растворе выполнялось равенствоаj = хj (для растворителя‚ ввиду10 = 1Ч‚ оно при этом также выполняется).
Часто используют также коэффициенты активности j = аj / хj ‚причем в последнем равенстве вместохj можно применять и другие меры концентрации.
Произвольность выбора активностей и коэффициентов активности в физической химии обычно связывают с проблемой выбора стандартного состояния‚ упомянутой в конце гл. 11. Даваемое при этом описание стандартного состояния может выглядеть очень странно (например‚ стандартное состояние‚ соответствующее выбору (12.2) для растворителя и (12.1) для растворенных веществ может быть описано как “бесконечно разбавленный раствор‚ в котором концентрации всех компонентов равны 1”). Мы рекомендуем при чтении соответствующей литературы не пытаться вообразить себе подобные состояния‚ а обращать внимание на формулы‚ определяющие j иаj.
12.2. Модель регулярного раствора. Вернемся к модели‚ рассмотренной в гл. 8‚ но вместо (8.1) примем
12 = (11 + 22)/2 + . (12.3)
В нулевом приближении не будем учитывать отклонений от хаотического расположения молекул двух сортов. При таком расположении среднее число соседей типа 2 у молекулы типа 1 будет zx2 , гдеz–—–полное число ближйших соседей (координационное число решетки). В результате раствор приобретет добавочную
энергию (и энтальпию) Низб–=–Nzx1x2 . Это даст
Ф = Фид + Nzx1x2 . (12.4)
Знак отклонений от идеальности (т.е. знак Физб = Ф–––Фид ) совпадает со знаком . Этот вывод сохраняется и в более точной теории‚ которую мы не будем рассматривать; ограничимся только качественным обсуждением влияния неаддитивности энергий на упорядоченность.
При отрицательных отклонениях от аддитивности ( < 0) расположение молекул двух сортов становится ближе к закономерному чередованию. Для твердого раствора типа замещения при х1 = х2 и достаточно большом –– должен образоваться вполне упорядоченный кристалл с чередующимися молекулами 1 и 2.
При положительном упорядочение проявится в увеличении среднего числа соседей типа 2 у молекулы типа 1 (и наоборот). В пределе два компонента разделятся.
В обоих случаях энтропия смешения будет уменьшена по сравнению с максимальным значением (8.9)‚ отвечающим вполне хаотическому расположению.
Остановимся более подробно на случае > 0. Рассматривая Ф как функцию х х1 при постоянном N‚ имеем:
/N =[x1Ч + (1 – x)2Ч] + kT[x ln x + (1 – x) ln (1 – x)] +[x (1 – x) z]. (12.5)
|
График функции Ф(х) построен на рис. 3а и 3б. Прямая 1 (аддитивная зависимость от— N1 и N2) изображает —выражение в первых квадратных скобках‚ кривая 2– —–второй (энтропийный) член‚ 3–—–их сумму Фид. Кривая— 4– представляет |
|
|
последний член в правой части (12.5)‚ т.е. Низб и Физб‚ а кривая 5‚ повторенная—в увеличенном масштабе на рис. 3б (где Ф1‚2 = N1‚2Ч)‚ — суммарную величину Ф. Кривая 5’ отвечает более высокой температуре (для нее рассчитаны кривые на рис.1 в главе 10).
Условие устойчивости раствора по отношению к распаду на фазы разной концентрации выражается неравенствами (6.11)‚ вместо которых удобно будет использовать равносильное (в силу соотношения Гиббса–—–Дюгема) неравенство
(2 /x2)T,p,N = [(1 – 2)/x ]T,p,N 0. (12.6)
(Величину
1– 2 = (2 /x2)T,p,N (12.7)
часто называют “химическим потенциалом раствора”.)
Условие (12.6) выполняется для идеального раствора (кривая 3 вогнутая)‚ а для неидеального оно выполнено‚ если вклад кривизны кривой 2–—–энтропийного вклада в Фид —– преобладает над вкладом кривизны кривой 4–—– вклада Физб. Однофазный раствор при этом устойчив. Так всегда будет при высоких температурах‚ но с пониженем температуры произойдет переход к обратной ситуации–—–на кривой Ф(х) появляется выпуклый участок (кривая 5). Температуру перехода‚ называемую критической температурой смешения Ткр‚ –нетрудно найти‚ если обратить внимание‚ что кривизна обеих кривых максимальна при х–=–1/2. Подставляя это значение‚ находим‚ что (12.6) обратится в равенство при
Т = Ткр = z /2k , х–=–хкр–=–1/2. (12.8)
Кривая 6 на рис. 3б построена для Т = Ткр.
В области‚ где выполнено неравенство‚ обратное (12.6) (участок между точками перегиба‚ отмеченными звездочками на кривой 5)‚ однофазное состояние является абсолютно неустойчивым‚ т.е. сколь угодно малое отклонение от равновесия может не убывать‚ как при отклонении от устойчивого состояния‚ а катастрофически возрастать. Для демонстрации этого достаточно рассмотреть какое-нибудь одно из возможных отклонений от равновесия. Пусть раствор находится в состоянии‚ изображаемом на рис. 1б кружком на кривой 5. Разобьем систему на две подсистемы A и Б‚ содержащие NA—и—NБ молекул‚—и—предположим‚ что концентрации в подсистемах изменились от х—до хA = х + xA и хБ = х + xБ. (Величины xA и xБ связаны вытекающим из законов сохранения хANA + хБNБ = хN и NA + NБ = N “правилом рычага”‚ ср. [1], §§ 101‚ 123;—на—рисунке—принято—для—простоты NA = NБ = N/2 и соответственно xA = – xБ – x.) Состояния подсистем изображаются точками A и Б. Суммарная величина свободной энтальпии в возмущенном состоянии‚ равная NAФA + NБФБ‚ изобразится точкой с абсциссой х‚ лежащей на соединяющей точки A и Б прямой 7. Видно‚ что возмущение снижает Ф‚ т.е оно выгодно; следовательно‚ состояние‚ изображенное кружком‚ неустойчиво. Возмущение будет нарастать до распада системы на две устойчивые фазы.
Наоборот‚ вне ограниченного звездочками участка‚ т.е. при выполнении (8)‚ отрезки‚ построенные аналогично 7‚ располагались бы выше кривой–5 — возмущение невыгодно‚ т.е. исходное состояние по отношению к нему устойчиво.
Достаточно очевидно‚ что это справедливо для любых возмущений‚ не выводящих точки A или Б за пределы области выполнения неравенства (12.8). Такие однофазные состояния устойчивы по отношению к любым малым возмущениям.
Устойчивые двухфазные состояния изобразятся прямой 8‚ касательной к кривой 5 в двух точках. Действительно‚ касательная—к—кривой—Ф(х)—имеет—угловой—коэффициент—–=–1–––2‚ а– значение— ординаты в точке—х—есть Ф–=–х1–+–(1–––х)2. Последнее равенство‚ таким образом‚ является также и уравнением касательной. Она отсекает на ординатах х = 0 и х = 1 отрезки‚ равные 2 и 1‚ и для двух точек‚ соединенных общей касательной‚ выполнены оба уравнения равновесия вида (6.9). Это–—–две фазы‚ на которые должен распасться раствор.
Отрезки кривой 5‚ лежащие между точками касания с прямой 8 и точками перегиба‚ изображают метастабильные однофазные состояния‚ которые переходят во вполне равновесные двухфазные состояния только путем образования зародышей и‚ следовательно‚ могут сохраняться длительное время.
Описанное поведение вполне подобно поведению газа Ван-дер-Ваальса: концентрация аналогична объему‚ химический потенциал–—–давлению.
|
На рис. 4 показан
вид изотерм (х)‚получаемых на основании (12.7)‚ и ход
равновесных изотерм в двухфазной
области. Положение последних находится
аналогично правилу Максвелла ([1]‚ §§
101‚ 112). (Заметим‚ что‚ хотя два вывода
правила Максвелла‚ приве-денные в
[1]‚ основаны‚ в конечном счете‚ на
втором начале термодина-мики‚ но
только второй из них может быть
непосредственно применен к
рассматриваемому здесь случаю.
Раз-личие состоит в том‚ что если Модель (12.5) определяет только форму изотерм (х); их относитель- смещение по оси обусловлено |
|
|
|
температурной зависимостью—1Ч—и—2Ч‚ которая здесь выбрана так‚ чтобы получить удобное расположе- |
|
Рис. 4. Изотермы (х) регулярного раствора. |

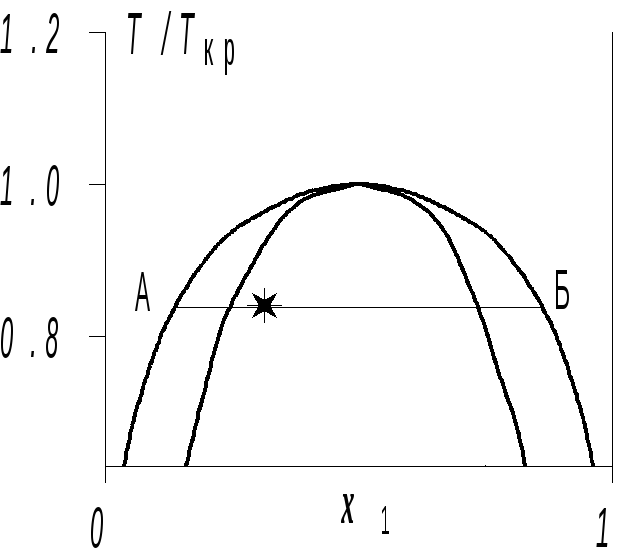
ние кривых на рисунке. Колоколообразные кривые ограничивают области равновесных двухфазных состояний и абсолютно неравновесных состояний. Обе кривые касаются между собой и с изотермой Т = Ткрв критической точке смешения. На рис. 5 эти же две кривые показаны в координатахх — Т. Концы горизонтального отрезка‚ проведен-ного в области несмешиваемости на уровнеТ/Ткр =0.84‚указывают составы фаз (A и Б)‚ на которые расслаивается раствор при этой температуре. Для общей концентрации‚ указанной звездочкой‚ хA/хБ = 3:1 (правило рычага).
Модель регулярного раствора позволяет качественно понять причины расслаивания растворов‚ но поведение реальных систем —обычно гораздо сложнее. В частности‚ относительное расположение областей— полной —смешиваемости— и
|
|
|
расслаивания бывает самым раз-личным. Существуют системы‚ смешиваю-щиеся при низких температурах и рас-слаивающиеся при высоких. Иногда область полной смешиваемости распола-гается между двумя областями рассла-ивания–—–низко- и высокотемпературной; встречаются системы‚—в которых замкнутая область несмеши-ваемости со всех сторон окружена на диаграмме х — Т областью——однофазных состояний. Примеры диаграмм различного вида приведены —в [1], § 124. Удовлетвори-тельные теории‚ позволяющие связать вид |
|
Рис. 5. Диаграмма состояний регулярного раствора (расслаивание). |
|
диаграмм состояния с особенностями меж-молекулярного взаимодействия в жидких растворах‚ существуют пока только для простейших случаев. |

Из причин‚ которые могут—приводить к отклонению свойств раствора от идеальности и не принимаются во внимание моделью регулярного раствора‚ можно назвать различие размеров между молекулами компонентов (напомним‚ кстати‚ что наглядное понятие “размера” не является. строгим: даже для сферически симметричной молекулы‚ такой как Ar, диаметром можно называть и равновесное расстояние между двумя изолированными молекулами‚ и межмолекулярное расстояние в кристалле‚ и среднее для данной температуры расстояние максимального сближения при столкновении). Другой важной причиной может быть наличие специфических взаимодействий‚ например‚ тенденции к соединению молекул A и В в группы определенного состава: AВ‚ AВ2‚ и т.д. ‚ Растворы с такой тенденцией называют ассоциированными‚ и рассматривают в первом приближении как идеальную смесь таких групп‚ концентрации которых определяют по законам химического равновесия (гл. 11). Заметим‚ что из такой модели следуютотрицательныеотклонения от идеальности‚ так как образование групп‚ как и любой процесс приближения к равновесию‚ должен вести к понижению Ф (гл. 5).
Л и т е р а т у р а
1. Сивухин Д.В.Общий курс физики. Т. 2. М.‚ 1990.
2. Фриш С.Э.‚ Тиморева A.В.Курс общей физики. Т. 1. М.‚1962
3. Толстой Н.A.Конспект лекций по физике. Л.‚ 1966.
4. Спартаков A.A.‚ Толстой Н.A.(составители). Методические указания по общему курсу физики (Некоторые вопросы термодинамики). Л.‚ 1990.
5. Сивухин Д.В.Общий курс физики. Т. 1. М.‚ 1974.
6. Сивухин Д.В.(редактор). Сборник задач по общему курсу физики. Термодинамика и молекулярная физика. М.‚ 1976.
7. Соловьев В.A.(составитель). Молекулярная физика жидкостей в курсе общей физики: метод. разработка. Л.‚ 1983