

Cycloaddition Reactions in Organic Synthesis
.pdf
|
|
|
References |
209 |
|
|
|
|
|
[24] |
Kobayashi, S.; Kusakabe, K.; Komiyama, |
[28] |
(a) Miyaura, N.; Ishiyama, T.; Sasaki, |
|
|
S.; Ishitani, H. J. Org. Chem. 1999, 64, |
|
H.; Ishikawa, M.; Satoh, M.; Suzuki, |
|
|
4220. |
|
A. J. Am. Chem. Soc. 1989, 111, 314; (b) |
|
[25] |
Ishitani, H.; Kitazawa, T.; Kobayashi, |
|
Cox, P. J.; Snieckus, V. Tetrahedron Lett. |
|
|
S. Tetrahedron Lett. 1999, 40, 2161. |
|
1992, 33, 2253; (c) Frenette, R.; Frie- |
|
[26] |
For chiral catalyst optimization using sol- |
|
sen, R. W. Tetrahedron Lett. 1994, 35, |
|
|
id-phase methods, see (a) Francis, M. B.; |
|
9177. |
|
|
Jacobsen, E. N. Angew. Chem., Int. Ed. |
[29] |
Kobayashi, S.; Akiyama, R.; Furuta, T.; |
|
|
Engl. 1999, 38, 937; (b) Porte, A. M.; Rei- |
|
Moriwaki, M. Molecules 1998, 2, 35. |
|
|
benspies, J.; Burgess, K. J. Am. Chem. |
[30] |
Kobayashi, S.; Kusakabe, K.; Ishitani, |
|
|
Soc. 1998, 120, 9180; (c) Gilbertson, |
|
H. Org. Lett. 2000, 2, 1225. |
|
|
S. R.; Wang, X. Tetrahedron Lett. 1996, 37, |
[31] |
Yao, S.; Saaby, S.; Hazell. R. G.; |
|
|
6475; (d) Shimizu, K. D.; Cole, B. M.; |
|
Jørgensen, K. A. Chem. Eur. J. 2000, 6, |
|
|
Krueger, C. A.; Kuntz, K. W.; Snapper, |
|
2435. |
|
|
M. L.; Hoveyda, A. H. Angew. Chem., Int. |
[32] |
Bromidge, S.; Wilson, P. C.; Whiting, |
|
|
Ed. Engl. 1997, 36, 1703, and references |
|
A. Tetrahedron Lett. 1998, 39, 8905. |
|
|
cited therein. |
[33] |
Jnoff, E.; Ghosez, L. J. Am. Chem. Soc. |
|
[27] |
Bayston, D. J.; Fraser, J. L.; Ashton, |
|
1999, 121, 2617. |
|
|
M. R. Baxter, A. D.; Polywka, M. E. C.; |
|
|
|
|
Moses, E. J. Org. Chem. 1998, 63, 3137. |
|
|
|

Cycloaddition Reactions in Organic Synthesis. 211
Edited by S. Kobayashi and K. A. Jorgensen
Copyright © 2001 Wiley-VCH Verlag GmbH
ISBNs: 3-527-30159-3 (Hardcover); 3-527-60025-6 (Electronic)
6
Asymmetric Metal-catalyzed 1,3-Dipolar Cycloaddition Reactions
Kurt Vesterager Gothelf
6.1
Introduction
The 1,3-dipolar cycloaddition reaction is the single most important method for the construction of heterocyclic five-membered rings in organic chemistry [1, 2]. Concerted cycloaddition reactions are also among the most powerful tools for the stereospecific creation of new chiral centers in organic molecules. When 1,2-disubstituted alkenes are involved in concerted 1,3-dipolar cycloaddition reactions, two new chiral centers on the alkene are formed in a stereospecific manner because of the syn attack on the double bond. This is shown for reactions of allyl anion-type and propargyl/ allenyl anion-type 1,3-dipoles in Scheme 6.1. Thus, the relative stereochemistry at C-4 and C-5 is always controlled by the geometric relationship of the substituents on the alkene for concerted 1,3-dipolar cycloaddition reactions [3, 4]. Depending on the structure of the dipole, up to four new contiguous chiral centers can be formed in the 1,3-dipolar cycloaddition reaction in a single step and the current challenge for 1,3-dipolar cycloaddition reactions is to control the absolute stereoselectivity of the reaction by the application of chiral metal catalysts.
Scheme 6.1
Asymmetric synthesis is a stimulating academic challenge, but since it has become clear that most chiral drugs can be administered safely only in the enantiomerically pure form, the industrial need for asymmetric methods has made research in asymmetric synthesis absolutely necessary [5]. This has driven a renaissance in the discipline of organic chemistry, because all of the old-established reactions need to be reinvestigated for their application in asymmetric synthesis [6]. This has also applied

212 6 Asymmetric Metal-catalyzed 1,3-Dipolar Cycloaddition Reactions
to the 1,3-dipolar cycloaddition reaction and during the past 15 years there has been enormous interest in asymmetric 1,3-dipolar cycloaddition reactions [7, 8]. Most of the research performed has, however, been on diastereoselective reactions that imply optically active substrates. Unlike the broad application of asymmetric catalysis in carboand hetero-Diels-Alder reactions, which has evolved since the mid-nine- teen-eighties [9], the use of enantioselective metal catalysts in asymmetric 1,3-dipolar cycloaddition reactions between alkenes and nitrones remained almost unexplored until 1993 [7]. The development of metal-catalyzed asymmetric 1,3-dipolar cycloaddition reactions that has been achieved up to 2000 is compiled in this chapter. After an introduction to the basics of metal-catalyzed 1,3-dipolar cycloaddition subsequent sections are divided according to the metal catalysts applied for the reactions.
6.2
Basic Aspects of Metal-catalyzed 1,3-Dipolar Cycloaddition Reactions
6.2.1
The 1,3-Dipoles
The 1,3-dipoles consist of elements from main groups IV, V, and VI. The parent 1,3-dipoles consist of elements from the second row and the central atom of the dipole is limited to N or O [10]. Thus, a limited number of structures can be formed by permutations of N, C, and O. If higher row elements are excluded twelve allyl anion type and six propargyl/allenyl anion type 1,3-dipoles can be obtained. However, metal-catalyzed asymmetric 1,3-dipolar cycloaddition reactions have only been explored for the five types of dipole shown in Scheme 6.2.
Scheme 6.2
Most studies in this field have been on nitrones. One of the reasons for this is probably because nitrones are readily available compounds that can be obtained from aldehydes, amines, imines, and oximes [2, 11]. Moreover, most acyclic ni-

6.2 Basic Aspects of Metal-catalyzed 1,3-Dipolar Cycloaddition Reactions 213
trones are stable compounds that can be stored under ambient conditions. Cyclic nitrones tend to be less stable, but there are also examples on the application of these. Azomethine ylides are unstable and have to be prepared in situ. Several methods have been developed for the synthesis of azomethine ylides, for example proton abstraction from imine derivatives of -amino acids, thermolysis or photolysis of aziridines and dehydrohalogenation of imonium salts [7, 12]. As with nitrones, azomethine ylides have found broad application in synthesis, but the number of reports on metal-catalyzed asymmetric reactions is very sparse. Carbonyl ylides are a less common type of 1,3-dipole, which have found only limited application in synthesis [13], although access to carbonyl ylides via rhodium carbenes has accelerated the development in this area [14, 15], and over the past three years the first examples of metal-catalyzed asymmetric reactions have appeared. Nitrile oxides on the other hand are, in close competition with nitrones, the most commonly applied 1,3-dipole for the synthesis of five-membered heterocyclic rings [2, 16]. They are easy available from aldoximes or primary nitro compounds, but most nitrile oxides must be prepared in situ, because of high reactivity and rapid dimerization. The high reactivity of nitrile oxides is probably one reason for the very few examples of catalytic control of this reaction [7]. Much attention has been devoted to the asymmetric reactions of alkenes with diazoalkanes, but the majority of the work is in relation to cyclopropanation chemistry, which will not be covered here [17]. The only example of a metal-catalyzed asymmetric reaction leading to a stable pyrazole product will be mentioned.
6.2.2
Frontier Molecular Orbital Interactions
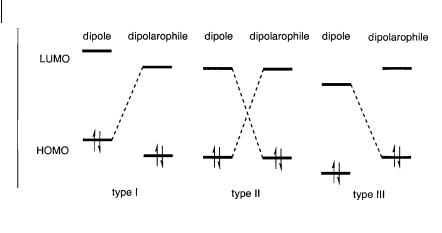
The transition state of the concerted 1,3-dipolar cycloaddition reaction is controlled by the frontier molecular orbitals (FMO) of the substrates. The LUMOdipole interacts with the HOMOalkene and the HOMOdipole interacts with the LUMOalkene [3, 18]. Sustman has classified 1,3-dipolar cycloaddition reactions into three types, based on the relative FMO energies between the dipole and the dipolarophile (Scheme 6.3) [19–21]. In type I reactions the dominant FMO interaction is that of the HOMOdipole with the LUMOdipolarophile. For type II reactions the similarity of the dipole and dipolarophile FMO energies implies that both HOMO-LUMO interactions are important. Cycloaddition reactions of type III are dominated by the interaction between the LUMOdipole and the HOMOdipolarophile.
Reactions of type I are typical for azomethine ylides and carbonyl ylides, whereas 1,3-dipolar cycloaddition reactions of nitrones are normally classified as type II [10]. Reactions of nitrile oxides are also classified as type II, but they are better classified as borderline to type III, since nitrile oxides have relatively low LUMO energies of –11 to –10 eV. It should be taken into account that the classification of a reaction is also dependent on the other reactant. Introduction of elec- tron-donating or electron-withdrawing substituents on the dipole or the dipolarophile can alter the relative FMO energies, and therefore the reaction type, dramatically [20, 21]. The reaction of N-methyl-C-phenylnitrone with methyl acrylate is
214 6 Asymmetric Metal-catalyzed 1,3-Dipolar Cycloaddition Reactions
Scheme 6.3
controlled by the HOMOdipole-LUMOdipolarophile interaction, whereas the reaction of the same nitrone with methyl vinyl ether is controlled by the LUMOdipole- HOMOdipolarophile interaction [11].
The relative FMO energies of the reagents are very important to the catalytic control of the reaction. Let us first have a look at nitrones where this principle has been explored in most detail [7, 22, 23]. To be able to control the stereochemistry of a reaction with a sub-stoichiometric amount of a ligand-metal catalyst it is desirable that large reaction rate accelerations are obtained, to assure that the reaction only takes place in the sphere of the metal and the chiral ligand. The strategy that was applied for the catalytic enhancement of the reaction rate has therefore been to alter the relative energies of the FMO of one of the substrates using chiral Lewis acid complexes [22]. This principle of activation can be applied to the 1,3-dipolar cycloaddition of nitrones with alkenes in two different ways. The reaction between a nitrone and an electron-deficient alkene such as an , -unsaturated carbonyl compound is primarily controlled by the interaction between HOMOnitrone-LUMOalkene (Scheme 6.4). By the application of a Lewis acid (LA) catalyst which is a strong electron acceptor it has been possible to decrease the energy of the FMOalkene via coordination of the enone to the Lewis acid. As a result of the decreased energy gap between the interacting FMO a rate acceleration of the reaction has been achieved [22].
The other catalytic approach to the 1,3-dipolar cycloaddition reaction is the “inverse electron-demand”, in which the nitrone is activated for addition to an electron-rich alkene such as e.g. a vinyl ether (Scheme 6.5). In this scenario the FMOalkene have higher energies than the FMOnitrone and the dominating interaction in the reaction will be LUMOnitrone-HOMOalkene. In the presence of a Lewis acid catalyst, the nitrone can coordinate to the catalyst, leading to a decrease of the LUMOnitrone energy. The decreased energy gap between the two FMOs responsible for the dominating interaction may lead to an enhanced rate of the 1,3-dipolar cycloaddition reaction. These principles of activation have proven to apply for the 1,3-dipolar cycloaddition reactions involving nitrones and in a single case also for diazoalkanes.

6.2 Basic Aspects of Metal-catalyzed 1,3-Dipolar Cycloaddition Reactions 215
Scheme 6.4
Scheme 6.5
For the reactions of other 1,3-dipoles, the catalyst-induced control of the enantioselectivity is achieved by other principles. Both for the metal-catalyzed reactions of azomethine ylides, carbonyl ylides and nitrile oxides the catalyst is crucial for the in situ formation of the 1,3-dipole from a precursor. After formation the 1,3-di- pole is coordinated to the catalyst because of a favored chelation and/or stabiliza-

216 6 Asymmetric Metal-catalyzed 1,3-Dipolar Cycloaddition Reactions
tion of the substrates, which apparently provide control of the enantioselectivity of the reaction.
6.2.3
The Selectivities of 1,3-Dipolar Cycloaddition Reactions
Three types of selectivity must be considered in 1,3-dipolar cycloaddition reactions – regioselectivity, diastereoselectivity, and enantioselectivity. The regioselectivity is controlled by both steric and electronic effects [18, 24]. For the addition to terminal alkenes the sterically most crowded functionality of the 1,3-dipole tends to add to the terminal carbon atom of the alkene, giving the 5-substituted isomer as shown for nitrones in Scheme 6.6. The steric effects may, however, be overruled by strong electronic effects [2]. In the cycloaddition reaction of electron-rich or electron-neutral alkenes with nitrones, the 5-substituted isomer is obtained. The reaction is primarily controlled by the LUMOdipole-HOMOdipolarophile interaction. The LUMOdipole has the largest coefficient at the carbon atom and the HOMOalkene has the largest coefficient at the terminal carbon atom. Thus, the nitrone and alkene combine in a regioselective manner to give the 5-isoxazolidine. This is obviously supported by steric factors. For terminal alkenes with an electron-withdrawing group, the reaction is primarily controlled by the HOMOdipole-LUMOdipolarophile interaction. The HOMOdipole has the largest coefficient at the oxygen atom, whereas the LUMOdipolarophile has the largest coefficient at the terminal carbon atom. This favors formation of the 4-isomer, but since steric factors oppose this, a mixture of regioisomers is often obtained [24]. However, in the reaction of nitrones with 1,2-disubstituted alkenes bearing an elec- tron-withdrawing group the steric factor is eliminated, leading to the FMO-con- trolled regioselectivity of the reaction with the 4-EWG-substituted isomer as the sole product (Scheme 6.6) [2, 11].
Scheme 6.6

6.2 Basic Aspects of Metal-catalyzed 1,3-Dipolar Cycloaddition Reactions 217
In the 1,3-dipolar cycloaddition reactions of especially allyl anion type 1,3-di- poles with alkenes the formation of diastereomers has to be considered. In reactions of nitrones with a terminal alkene the nitrone can approach the alkene in an endo or an exo fashion giving rise to two different diastereomers. The nomenclature endo and exo is well known from the Diels-Alder reaction [3]. The endo isomer arises from the reaction in which the nitrogen atom of the dipole points in the same direction as the substituent of the alkene as outlined in Scheme 6.7. However, compared with the Diels-Alder reaction in which the endo transition state is stabilized by secondary -orbital interactions, the actual interaction of the N-nitrone pz-orbital with a vicinal pz-orbital on the alkene, and thus the stabilization, is small [25]. The endo/exo selectivity in the 1,3-dipolar cycloaddition reaction is therefore primarily controlled by the structure of the substrates or by a catalyst. It should be noticed that for reactions in which the nitrone can undergo Z/E-inter- conversion, the endo/exo assignment of the products is misleading and therefore cis or trans should be used instead.
Scheme 6.7
For azomethine ylides and carbonyl ylides, the diastereoselectivity is more complex as the presence of an additional chiral center in the product allows for the formation of four diastereomers. Since the few reactions that are described in this chapter of these dipoles give rise to only one diastereomer, this topic will not be mentioned further here [10].
Finally, there is the enantioselectivity of the 1,3-dipolar cycloaddition reactions. This chapter is limited to describing only the metal-catalyzed asymmetric 1,3-dipo- lar cycloaddition reactions that involve non-chiral starting materials. The only fac-

218 6 Asymmetric Metal-catalyzed 1,3-Dipolar Cycloaddition Reactions
tor present for control of the enantioselectivity is therefore the chiral catalyst. The use of chiral metal-ligand complexes to control the enantioselectivity of 1,3-dipolar cycloaddition reactions is the primary focus of this chapter. Therefore the chapter has been divided into sections based on the metal catalysts.
6.3
Boron Catalysts for Reactions of Nitrones
Scheeren et al. reported the first enantioselective metal-catalyzed 1,3-dipolar cycloaddition reaction of nitrones with alkenes in 1994 [26]. Their approach involved C,N-diphenylnitrone 1a and ketene acetals 2, in the presence of the amino acidderived oxazaborolidinones 3 as the catalyst (Scheme 6.8). This type of boron catalyst has been used successfully for asymmetric Diels-Alder reactions [27, 28]. In this reaction the nitrone is activated, according to the inverse electron-demand, for a 1,3-dipolar cycloaddition with the electron-rich alkene. The reaction is thus controlled by the LUMOnitrone-HOMOalkene interaction. They found that coordination of the nitrone to the boron Lewis acid strongly accelerated the 1,3-dipolar cycloaddition reaction with ketene acetals. The reactions of 1a with 2a,b, catalyzed by 20 mol% of oxazaborolidinones such as 3a,b were carried out at –78 C. In some reactions fair enantioselectivities were induced by the catalysts, thus, 4a was obtained with an optical purity of 74% ee, however, in a low yield. The reaction involving 2b gave the C-3, C-4-cis isomer 4b as the only diastereomer of the product with 62% ee.
Scheme 6.8
In an extension of this work Scheeren et al. studied a series of derivatives of N-to- syl-oxazaborolidinones as catalysts for the 1,3-dipolar cycloaddition reaction of 1 with 2b [29]. The addition of a co-solvent appeared to be of major importance. Catalyst 3b was synthesized from the corresponding amino acid and BH3·THF, hence, THF was present as a co-solvent. In this reaction (–)-4b was obtained with 62% ee. If the catalyst instead was synthesized from the amino acid and

6.4 Aluminum Catalysts for Reactions of Nitrones 219
BH3 · SMe2, and diphenyl ether was added, a remarkable reversal of the enantioselectivity of the reaction occurred, since (+)-4b was now obtained as the major isomer. Furthermore, the ee in this approach was improved to be 79%.
In a more recent work the same research group has applied cyclic and acyclic vinyl ethers in the oxazaborolidinone-catalyzed 1,3-dipolar cycloaddition reaction with nitrones [30]. The reaction between nitrone 5 and 2,3-dihydrofuran 6 with 20 mol% of the phenyl glycine-derived catalyst 3c, gave the product 7 in 56% yield as the sole diastereomer, however, with a low ee of 38% (Scheme 6.9).
Scheme 6.9
In an analogous study by Meske, the impact of various oxazaborolidinone catalysts for the 1,3-dipolar cycloaddition reactions between acyclic nitrones and vinyl ethers was studied [31]. Both the diastereoand the enantioselectivities obtained in this work were low. The highest enantioselectivity was obtained by the application of 100 mol% of the tert-butyl-substituted oxazaborolidinone catalyst 3d [27, 32] in the 1,3-dipolar cycloaddition reaction between nitrone 1a and ethyl vinyl ether 8a giving endo-9a and exo-9a in 42% and 27% isolated yield, respectively, with up to 20% ee for endo-9a as the best result (Scheme 6.10).
Scheme 6.10
6.4
Aluminum Catalysts for Reactions of Nitrones
As for boron catalysts, the aluminum catalysts have exclusively been applied for the inverse electron-demand 1,3-dipolar cycloaddition between alkenes and nitrones. The first contribution to this field was published by Jørgensen et al. in