

Cycloaddition Reactions in Organic Synthesis
.pdf7.4 Diazo Cycloadditions 281
wis acid catalyst which is rather insoluble in the reaction solvent, the more acidic methine hydrogen migrates to give Nc.
It was our delight that the reactions catalyzed were activated even at –40 C in the presence of a catalytic amount of achiral titanium catalysts (10 mol%) to afford the desilylacetylated 2-pyrazoline cycloadduct Na, 1-acetyl-4-methyl-5-(2-oxo-3- oxazolidinylcarbonyl)-2-pyrazoline, in high yields as the far major product (Scheme 7.35). Although some chiral titanium TADDOlate catalysts were successfully applied to activate these reactions leading to the moderate enantioselectivities (up to 55% ee), the chemical yields were not satisfactory.
TiBr2(OPr-i)2
TiBr2(OPr-i)2/MS 4A
TADDOL/Ph/TiBr2 (OPr- )2
TADDOL/1-Np/TiBr2 (OPr- )2
TADDOL/2-Np/TiBr2 (OPr- )2
TADDOL/Ph/TiBr2 (OPr- )2/MS 4A
TADDOL/Ph/TiBr2 (OPr- )2/MS 4A
Scheme 7.35
7.4.2
Zinc Complex-catalyzed Asymmetric Reactions
After screening of several of other chiral catalysts, the nickel(II) and zinc(II) complexes of R,R-DBFOX/Ph ligand were found to be reactive even when they were used in catalytic amounts (10 mol%) in dichloromethane. A high chemical yield of the desilylacetylated 2-pyrazoline cycloadduct Na was obtained as the single product, and the zinc(II) complex was found to be the best catalyst [81]. Thus, the maximum enantioselectivity as high as 99% ee was attained in the reaction catalyzed by R,R-DBFOX/Ph·Zn(ClO4)2·3H2O (10 mol%) at –40 C among a little excess of trimethylsilyldiazomethane (1.2 equiv.), 3-crotonoyl-2-oxazolidinone, acetic anhydride (1.1 equiv.), and MS 4 Å (Scheme 7.36). The presence of MS 4 Å was essential because no formation of any cycloadducts was observed without MS 4 Å, the starting trimethylsilyldiazomethane being recovered intact. The role of MS 4 Å is simply a dehydrating agent in this case since the comparable results were obtained in the reaction catalyzed by the anhydrous complex catalyst prepared from R,R-DBFOX/Ph, ZnI2, and AgClO4.
Unfortunately the reaction of trimethylsilyldiazomethane with 2-acryloyl-2-oxa- zolidinone led to a racemic result. Since 2-acryloyl-2-oxazolidinone has a terminal-

282 7 Aqua Complex Lewis Acid Catalysts for Asymmetric 3+2 Cycloaddition Reactions
Scheme 7.36
ly unsubstituted reaction site, its reaction presumably proceeded through a different reaction mechanism, either the endo approach of trimethylsilyldiazomethane or stepwise linear approach. Surprisingly, both the 3-acryloyl-2-oxazolidinones bearing propyl and isopropyl substituents at the -position were much less enantioselective than the methyl-substituted dipolarophile (Scheme 7.37). Especially, the reaction of 3-(2-hexenoyl)-2-oxazolidinone as the primary alkyl-substituted dipolarophile never exceeded an enantioselectivity of 50% ee. These -substituents, isopropyl and propyl moieties, have higher mobility than the methyl substituent, and therefore, some serious steric hindrance should exist against one of the shielding phenyl groups so that the reaction site departs from the shielding phenyl group. As a result, efficiency of the chiral shielding became rather ineffective.
On the other hand, use of 4,4-dimethyl-2-oxazolidinone as chiral auxiliary was very effective. It was found that the R,R-DBFOX/Ph·Mg(ClO4)2 complex was the catalyst of choice to mediate the reactions with 3-crotonoyl-4,4-dimethyl-2-oxazoli- dinone, because both the R,R-DBFOX/Ph-zinc(II) and –nickel(II) complexes were totally inactive. Thus, the R,R-DBFOX/Ph·Mg(ClO4)2-catalyzed reaction of trimethylsilyldiazomethane with 3-crotonoyl-4,4-dimethyl-2-oxazolidinone in the presence of MS 4 Å proceeded smoothly even at –78 C to give the corresponding cycloadduct in 75% yield with the enantioselectivity of 97% ee. Other dipolarophiles such as the 4,4-dimethyl-2-oxazolidinones having 2-hexenoyl and 4-methyl- 2-pentenoyl substituents at the nitrogen atom of the oxazolidinone-chelating auxiliary showed similarly high enantioselectivities of 98% ee regardless of the -sub- stituents of dipolarophiles (Scheme 7.38).

7.4 Diazo Cycloadditions 283
Scheme 7.37
Scheme 7.38
7.4.3
Transition Structures
The desilylacetylated cycloadducts, produced from the reactions of trimethylsilyldiazomethane with 3-crotonoyl-2-oxazolidinone or 3-crotonoyl-4,4-dimethyl-2-oxa- zolidinone, were transformed to methyl trans-1-acetyl-4-methyl-1-pyrazoline-5-car- boxylate through the reactions with dimethoxymagnesium at –20 C. When the optical rotations and chiral HPLC data were compared between these two esters, it was found that these two products had opposite absolute stereochemistry (Scheme 7.39). The absolute configuration was identified on the basis of the X- ray-determined structure of the major diastereomer of cycloadduct derived from the reaction of trimethylsilyldiazomethane to (S)-3-crotonoyl-4-methyl-2-oxazolidi- none.
The cycloaddition product derived from 3-crotonoyl-2-oxazolidinone was identified to be the 4S,5R-enantiomer of 2-pyrazoline cycloadduct, meaning that the re,si-enan-

284 7 Aqua Complex Lewis Acid Catalysts for Asymmetric 3+2 Cycloaddition Reactions
Scheme 7.39
tioface of the dipolarophile was involved in the attack of trimethylsilyldiazomethane as a result of the chiral shielding from the top 4-phenyl substituent, as shown in the trigonal bipyramid transition structure TS-O (Scheme 7.40). The selected enantioface of 3-crotonoyl-2-oxazolidinone was the same to that involved in the transition structure of the R,R-DBFOX/Ph·Ni(ClO4)2·3H2O-catalyzed Diels-Alder reaction using cyclopentadiene and the same dienophile [39]. Consequently, the absolute configuration of the cycloaddition product produced in the diazo cycloaddition reaction of 3-crotonoyl-4,4-dimethyl-2-oxazolidinone was the 4R,5S enantiomer which resulted from the selection of si,re-enantioface of the oxazolidinone dipolarophile. The twisted trigonal bipyramid type transition structure TS-P is most likely to be involved if this dipolarophile has worked as bidentate dipolarophile in the reaction, in which the absolute configuration of the cycloadduct was induced from the chiral shielding by the opposite phenyl shielding substituent (the bottom 4-phenyl).
Scheme 7.40

7.5 Conjugate Additions 285
However, we have so far no satisfactory explanation for the phenomenon that the reaction was selectively activated only by the magnesium complex catalyst. Since two methyl substituents exist at the 4-position of oxazolidinone-chelating auxiliary, the both enantiofaces of the reacting substrate complex should be sterically hindered. This is no doubt the major reason for the failure of activation by the nickel(II) and zinc(II) complex catalysts in the trigonal bipyramid transition structure like TS-O. The R,R-DBFOX/Ph-magnesium complex was apparently not much better catalyst in the reactions of trimethylsilyldiazomethane with 3-crotonoyl-2-ox- azolidinone, so the reaction through the trigonal bipyramid transition structure, than the corresponding zinc(II) and nickel(II) catalysts. Therefore, one possibility would be the magnesium complex is especially an active catalyst in the twisted trigonal bipyramid transition structure. Activation through the single point coordination of dipolarophile also remains as a possibility.
7.5
Conjugate Additions
One of the remarkable features of the R,R-DBFOX/Ph-transition metal aqua complexes is not only high enantioselectivity in the reactions catalyzed by the complexes but also high stability and tolerance toward nucleophilic or coordinating reagents. Therefore, these aqua complexes can be utilized as tolerant chiral Lewis acid catalysts in the catalyzed enantioselective reactions using highly coordinating nucleophiles, which are otherwise difficult to achieve [39, 82]. Probably the water ligands are rapidly exchanged with the acceptor and donor molecules, and this rapid ligand exchange reaction should be responsible for the high catalytic activity of these aqua complexes [57]. The aforementioned successful applications to the catalyzed enantioselective 1,3-dipolar cycloaddition reactions of nitrones [68, 76], cyclic nitronates [75], and diazoalkanes [81] are the examples. Accordingly, we further investigated the Lewis acid-catalyzed conjugate addition reactions using thiols [83], hydroxylamines, and carbon nucleophiles in the presence of amine bases.
7.5.1
Thiol Conjugate Additions
Quite a number of asymmetric thiol conjugate addition reactions are known [84], but previous examples of enantioselective thiol conjugate additions were based on the activation of thiol nucleophiles by use of chiral base catalysts such as amino alcohols [85], the lithium thiolate complex of amino bisether [86], and a lanthanide tris(binaphthoxide) [87]. No examples have been reported for the enantioselective thiol conjugate additions through the activation of acceptors by the aid of chiral Lewis acid catalysts. We therefore focussed on the potential of R,R-DBFOX/ Ph aqua complex catalysts as highly tolerant chiral Lewis acid catalyst in thiol conjugate addition reactions.

286 7 Aqua Complex Lewis Acid Catalysts for Asymmetric 3+2 Cycloaddition Reactions
Among a variety of DBFOX/Ph complexes examined as chiral catalysts in thiol conjugate addition reactions between thiophenol and 3-crotonoyl-2-oxazolidinone, the nickel(II) aqua complex R,R-DBFOX/Ph·Ni(ClO4)2·3H2O was exceptionally effective (Scheme 7.41) [83]. Although the magnesium and zinc complexes prepared from R,R-DBFOX/Ph ligand by treatment with Mg(ClO4)2, Zn(ClO4)2·6H2O, Zn(OTf)2, or ZnI2 showed satisfactory catalytic activity, the enantioselectivities observed in the catalyzed thiol conjugate additions were relatively poor. On the other hand, metal complexes prepared from the perchlorates of copper(II), iron(II), and manganese(II) ions showed only a low catalytic activity. Accordingly, reactions of a variety of thiols were catalyzed by the aqua nickel(II) complex R,R-DBFOX/ Ph·Ni(ClO4)2·3H2O to give the corresponding adducts. Satisfactorily high enantioselectivities as well as high chemical yields were observed with some exceptions when the reactions were performed in THF at room temperature. Based on the absolute configuration of the thiophenol adduct, it was found that the thiol conjugate addition took place on the si face of the 3-crotonoyl-2-oxazolidinone.
Scheme 7.41
Enantioselectivities were found to change sharply depending upon the reaction conditions including catalyst structure, reaction temperature, solvent, and additives. Some representative examples of such selectivity dependence are listed in Scheme 7.42. The thiol adduct was formed with 79% ee (81% yield) when the reaction was catalyzed by the R,R-DBFOX/Ph aqua nickel(II) complex at room temperature in dichloromethane. Reactions using either the anhydrous complex or the aqua complex with MS 4 Å gave a racemic adduct, however, indicating that the aqua complex should be more favored than the anhydrous complex in thiol conjugate additions. Slow addition of thiophenol to the dichloromethane solution of 3-crotonoyl- 2-oxazolidinone was ineffective for enantioselectivity. Enantioselectivity was dramatically lowered and reversed to –17% ee in the reaction at –78 C. A similar tendency was observed in the reactions in diethyl ether and THF. For example, a satisfactory enantioselectivity (80% ee) was observed in the reaction in THF at room temperature, while the selectivity almost disappeared (7% ee) at 0 C.

7.5 Conjugate Additions 287
Scheme 7.42
To examine such high sensitivity of enantioselectivity to the reaction conditions, the reactions of thiophenol with 3-crotonoyl-2-oxazolidinone were performed in dichloromethane at room temperature in the presence of a variety of additives. Although addition of methanol (CH2Cl2–MeOH = 10 : 1 v/v) did not affect either the chemical yield or enantioselectivity (quant, 82% ee), addition of acetonitrile or N,N-dimethylformamide (both 1 : 1 v/v ratios) slowed the reactions (13%, 15% yields) and provided products with lower enantioselectivities (19%, 30% ee). The presence of acetic acid, even in a small amount (CH2Cl2–AcOH = 10 : 1 v/v), gave the racemic product, while saturated aqueous ammonium chloride provided a reversed enantioselectivity (CH2Cl2–sat. NH4Cl aq. = 10 : 1 v/v, 99% yield, –27% ee). To our delight, however, reaction in the mixed solvent CH2Cl2–THF = 10 : 1 v/v catalyzed by the R,R-DBFOX/Ph aqua nickel(II) complex at 0 C in the presence of N,N,N ,N -tetramethyl-1,8-diaminonaphthalene (proton sponge, 10 mol%) gave the best result (84% yield, 94% ee). Some other thiols provided excellent enantioselectivities under similar reaction conditions with 97% ee for a bulky thiol such as o-isopropylbenzenethiol (Scheme 7.43).
At the beginning of the work, it was suspected that thiols would strongly coordinate to the Lewis acid catalyst to poison its catalytic activity even if the R,R- DBFOX/Ph·Ni(ClO4)2·3H2O catalyst is tolerant. We therefore examined the interaction between thiophenol and the catalyst R,R-DBFOX/Ph·Ni(ClO4)2·3H2O to learn about the catalytic activity of the thiol-coordinating complex. When thiophenol was added to the solution of R,R-DBFOX/Ph·Ni(ClO4)2·3H2O in THF, the original pale blue color of the catalyst gradually faded to reddish brown. This color change was rapid in dichloromethane, probably arising from the coordination of thiol to the catalyst. A brown colored solid was isolated as precipitate on treatment with a mixture of isopropyl alcohol and hexane, and this solid was

288 7 Aqua Complex Lewis Acid Catalysts for Asymmetric 3+2 Cycloaddition Reactions
Scheme 7.43
found to show sufficient catalytic activity in the reaction leading to a high enantioselectivity (97% yield, 70% ee). Accordingly, it seems likely that the thiol certainly binds with the catalyst, but the binding is not so strong that the thiol ligand may be easily replaced with the acceptor molecule in the reaction. This ligand exchange should be more favored in a coordinating media such as THF. However at the same time, THF competes with the acceptor molecule in coordination to the catalyst to deactivate the reaction. In the presence of an amine base such as pyridine or triethylamine, a totally inert reddish brown complex immediately precipitated. Since the resulting brown solid is totally insoluble in the reaction medium and free from perchlorate ions (according to analysis for chloride), we assume that the perchlorate counter-ions have been replaced with the highly nucleophilic thiolate ions.
The time-dependence of enantioselectivity in the reaction thiophenol with 3-cro- tonoyl-2-oxazolidinone catalyzed by R,R-DBFOX/Ph·Ni(ClO4)2·3H2O at room temperature in THF is shown in Scheme 7.44. After 3 h, the yield of the thiol adduct is 70% with the enantioselectivity of 91% ee, but the enantioselectivity was 80% ee at the completion of reaction after 24 h (yield 100%). Although the catalyst maintains a high catalytic activity, and hence a satisfactory enantioselectivity, at the early stage of reaction, the deterioration of catalyst cannot be neglected thereafter even under neutral conditions.
7.5.2
Hydroxylamine Conjugate Additions
With the success in Lewis acid-catalyzed thiol conjugate addition reactions mentioned above, we further tried to apply the R,R-DBFOX/Ph-nickel(II) aqua complex catalyst to the catalyzed asymmetric conjugate addition reactions of hydroxylamines [88, 89]. However, after some preliminary examinations, we found that

7.5 Conjugate Additions 289
Scheme 7.44
these catalysts were totally ineffective in the reactions of O-benzylhydroxylamine and N-benzylhydroxylamine with 3-(2-alkenoyl)-2-oxazolidinones. Both rate acceleration and enantioselectivities were disappointingly low.
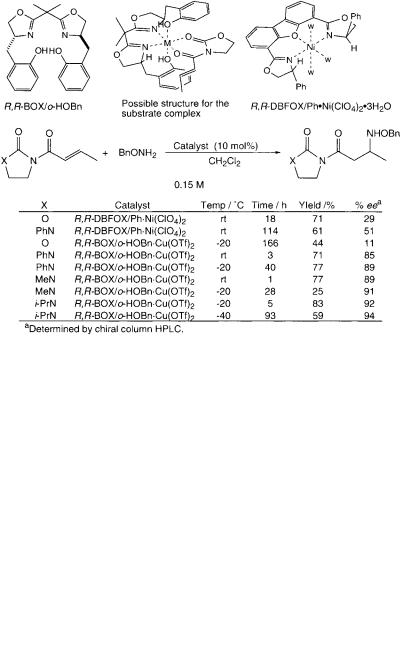
We therefore prepared a new chiral ligand, (R,R)-isopropylidene-2,2 -bis[4-(o-hy- droxybenzyl)oxazoline)], hereafter designated R,R-BOX/o-HOBn. To our delight, the copper(II) complex catalyst prepared from R,R-BOX/o-HOBn ligand and Cu(OTf)2 was quite effective (Scheme 7.45). Especially, the reaction of O-benzylhy- droxylamine with 1-crotonoyl-3-isopropyl-2-imidazolidinone in dichloromethane (0.15 m) at –40 C in the presence of R,R-BOX/o-HOBn·Cu(OTf)2 (10 mol%) provided the maximum enantioselectivity of 94% ee.
The importance of the o-hydroxyl moiety of the 4-benzyl-shielding group of R,R- BOX/o-HOBn·Cu(OTf)2 complex was indicated when enantioselectivities were compared between the following two reactions. Thus, the enantioselectivity observed in the reaction of O-benzylhydroxylamine with 1-crotonoyl-3-phenyl-2-imi- dazolidinone catalyzed by this catalyst was 85% ee, while that observed in a similar reaction catalyzed by R,R-BOX/Bn.Cu(OTf)2 having no hydroxyl moiety was much lower (71% ee). In these reactions, the same mode of chirality was induced (Scheme 7.46). We believe the free hydroxyl groups can weakly coordinate to the copper(II) ion to hinder the free rotation of the benzyl-shielding substituent across the C(4)–CH2 bond. This conformational lock would either make the coordination of acceptor molecules to the metallic center of catalyst easy or increase the efficiency of chiral shielding of the coordinated acceptor molecules.
1-Alkyl-2-imidazolidinones-chelating auxiliaries are more electron donating than 1-phenyl-2-imidazolidinone and 2-oxazolidinone auxiliaries so that the unsaturated amides of 1-alkyl-2-imidazolidinones should be less reactive under uncatalyzed conditions than those of 3-phenyl-2-imidazolidinone and 2-oxazolidinone. On the other hand, the coordinating ability to the catalyst is increased to be activated. Accordingly, the reaction rate difference between the catalyzed and uncatalyzed reac-
290 7 Aqua Complex Lewis Acid Catalysts for Asymmetric 3+2 Cycloaddition Reactions
Scheme 7.45
Scheme 7.46
tions should be greater in the case of derivatives of 1-alkyl-2-imidazolidinones. Competitive coordination of substrates, this case O-benzylhydroxylamine and acceptors, affects the total reaction rate in the Lewis acid-catalyzed reactions using