

Cycloaddition Reactions in Organic Synthesis
.pdf230 6 Asymmetric Metal-catalyzed 1,3-Dipolar Cycloaddition Reactions
reaction between nitrone 1a and alkene 19a led to endo/exo ratios between 18:82 and 8:92 and enantioselectivities of up to 56% ee (Scheme 6.24). The enantioselectivities are thus slightly decreased compared to the similar reactions of the homogeneous catalysts [56, 68]. They also made a study of the relationship between the enantiomeric purity of the ligand of the homogeneous catalyst 29, and the products obtained in both the 1,3-dipolar cycloaddition reaction between 1a and 19a and in the Diels-Alder reaction of 19a with cyclopentadiene. Surprisingly, the 1,3- dipolar cycloaddition shows a linear relationship, whereas the Diels-Alder reaction shows a positive non-linear relationship. In a recent work Seebach et al. have studied the use and re-use of Ti(OTs)2-TADDOLate catalysts immobilized on porous silica gel [69]. The selectivity obtained in the 1,3-dipolar cycloaddition between nitrone 1a and 19a catalyzed by 30, was only slightly lower compared to the corresponding homogeneous reaction [65]. The same batch of the ligand in 30 could be used in four consecutive reactions with no significant loss of activity, when the ligand was carefully washed between the reactions [69].
Scheme 6.24
6.6 Titanium Catalysts for Reactions of Nitrones and Diazoalkanes 231
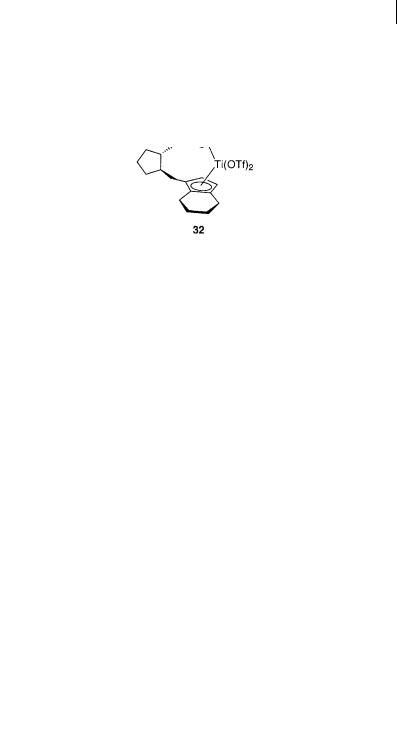
A quite different type of titanium catalyst has been used in an inverse electron-de- mand 1,3-dipolar cycloaddition. Bosnich et al. applied the chiral titanocene-(OTf)2 complex 32 for the 1,3-dipolar cycloaddition between the cyclic nitrone 14a and the ketene acetal 2c (Scheme 6.25). The reaction only proceeded in the presence of the catalyst and a good cis/trans ratio of 8:92 was obtained using catalyst 32, however, only 14% ee was observed for the major isomer [70].
Scheme 6.25
The normal electron-demand principle of activation of 1,3-dipolar cycloaddition reactions of nitrones has also been tested for the 1,3-dipolar cycloaddition reaction of alkenes with diazoalkanes [71]. The reaction of ethyl diazoacetate 33 with 19b in the presence of a TiCl2-TADDOLate catalyst 23a afforded the 1,3-dipolar cycloaddition product 34 in good yield and with 30–40% ee (Scheme 6.26).
Scheme 6.26

232 6 Asymmetric Metal-catalyzed 1,3-Dipolar Cycloaddition Reactions
6.7
Nickel Catalysts for Reactions of Nitrones
In 1998 Kanemasa et al. published the structure of a new dibenzofuranyl 2,2 -bis- oxazoline (DBFOX) which has proved to be an excellent ligand for a variety of Lewis acids [72]. The catalytic reactions that have been developed using this type of Lewis acid-DBFOX complexes, which include some new important catalytic 1,3-di- polar cycloaddition reactions of nitrones, nitronates and diazo compounds are described in a separate chapter in this book. Thus, in this section only the work on 1,3-dipolar cycloaddition reactions that has been published at the present will be described [73]. In this work a Ni(ClO4)2-PhDBFOX complex 35 is applied as the chiral catalyst (Scheme 6.27). Quite remarkably this Lewis acid catalyst can be formed from the aqueous Ni(ClO4)2 · 6H2O salt and the ligand. The water could be removed by the addition of MS 4 Å. The reaction between different nitrones 1 and crotonoyloxazolidinone 19a proceeded in the presence of 10 mol% of the dicationic nickel complex 35 as the catalyst. Although long reaction times were required to obtain good yields, the reactions proceeded, in most cases, with very high endo selectivities and, in several cases, > 99% ee of the endo products 21 was obtained. So far this catalyst is undoubtedly the most selective catalyst for the normal electron-demand 1,3-dipolar cycloaddition reaction between nitrones and alkenes, especially, with respect to the enantioselectivity of the reaction [73].
Scheme 6.27

6.8 Copper Catalysts for Reactions of Nitrones 233
6.8
Copper Catalysts for Reactions of Nitrones
The enantioselective inverse electron-demand 1,3-dipolar cycloaddition reactions of nitrones with alkenes described so far were catalyzed by metal complexes that favor a monodentate coordination of the nitrone, such as boron and aluminum complexes. However, the glyoxylate-derived nitrone 36 favors a bidentate coordination to the catalyst. This nitrone is a very interesting substrate, since the products that are obtained from the reaction with alkenes are masked -amino acids. One of the characteristics of nitrones such as 36, having an ester moiety in the position, is the swift E/Z equilibrium at room temperature (Scheme 6.28). In the crystalline form nitrone 36 exists as the pure Z isomer, however, in solution nitrone 36 have been shown to exists as a mixture of the E and Z isomers. This equilibrium could however be shifted to the Z isomer in the presence of a Lewis acid [74].
Scheme 6.28
To control the stereochemistry of 1,3-dipolar cycloaddition reactions involving this type of nitrone, chiral copper catalysts were applied [74]. For the 1,3-dipolar cycloaddition reaction of 36 was chosen the electron-rich ethyl vinyl ether 8a as the dipolarophile (Scheme 6.29). In the absence of a catalyst the reaction of 36 with 8a in CH2Cl2 gave a low conversion of 40% after 66 h and a 24 : 76 mixture of exo- 38a and endo-38a was obtained. A series of chiral catalysts was investigated for the reaction and the Cu(OTf)2-BOX complex 37a was found to be the most suitable catalyst for this reaction. In the presence of 25 mol% of 37a the reaction proceeded at rt in CH2Cl2 to give a conversion of 98% after only 8.5 h. The diastereoselectivity of the reaction was improved to an exo/endo ratio of 84 : 16 and, as the most significant result, exo-38a was obtained with 89% ee. By changing the solvent to toluene the diastereoselectivity of the reaction was slightly lowered, but the enantioselectivity was improved to 93% ee [74].
A model for the intermediate consisting of substrates 36 and 8a coordinated to catalyst 37a was proposed as shown in Scheme 6.30 [74]. In the model 39 the two triflate ligands are dissociated from copper. The ligands are arranged around copper as a trigonal bipyramid and it should be noted that in this model the oxygen atom of the vinyl ether 8a also coordinates to the metal center. However, another tetrahedral intermediate consisting of only the catalyst and the nitrone could also account for the absolute selectivity of the reaction.
Another rather peculiar asymmetric copper-catalyzed reaction was published some years earlier by Miura et al. [75]. The reaction is outlined in Scheme 6.31
234 6 Asymmetric Metal-catalyzed 1,3-Dipolar Cycloaddition Reactions
Scheme 6.29
Scheme 6.30
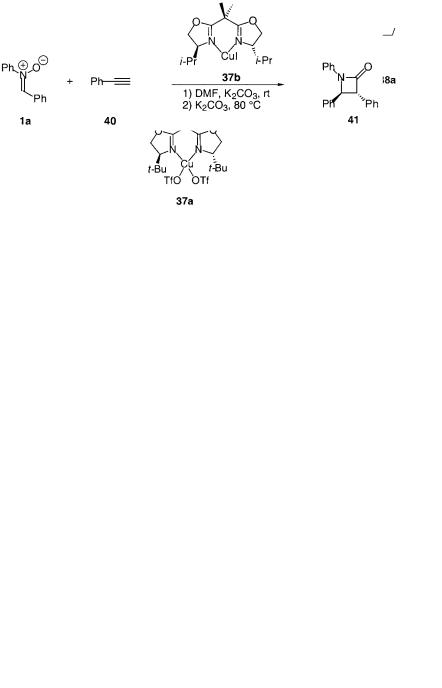
and it involves the reaction of nitrone 1a with phenylacetylene 40, catalyzed by CuI–i-Pr–BOX 37b. The product of this reaction is not a 1,3-dipolar cycloaddition adduct, rather it is the azetidone 41. By using 1 equivalent of the chiral catalyst the trans isomer 41 is obtained in 54 yield with 68% ee. If the catalyst loading is lowered to 10 mol% CuI and 20 mol% ligand the selectivity decreases to 57% ee.
Scheme 6.31

6.9 Zinc Catalysts for Reactions of Nitrones and Nitrile Oxides 235
A mechanism for this reaction has been proposed [75]. The first key intermediate in the reaction is the copper(I) acetylide 42. The additional ligand may be solvent or H2O. The acetylene moiety in 42 is activated for a 1,3-dipolar cycloaddition with the nitrone to give intermediate 43, with introduction of chirality in the product. A possible route to cis/trans-41 might be via intermediate 44. Finally, the cis isomer is isomerized into the thermally more stable trans-41. It should be mentioned that the mechanism outlined in Scheme 6.32 was originally proposed for a racemic version of the reaction to which water was added.
Scheme 6.32
6.9
Zinc Catalysts for Reactions of Nitrones and Nitrile Oxides
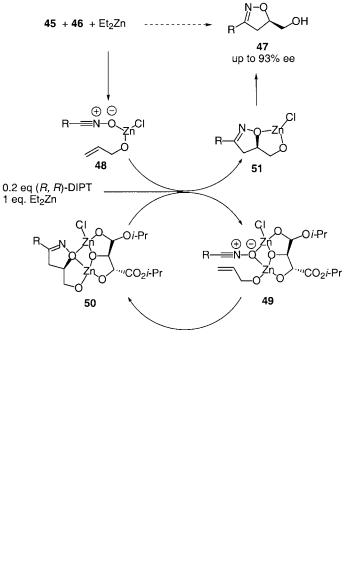
The first, and so far only, metal-catalyzed asymmetric 1,3-dipolar cycloaddition reaction of nitrile oxides with alkenes was reported by Ukaji et al. [76, 77]. Upon treatment of allyl alcohol 45 with diethylzinc and (R,R)-diisopropyltartrate, followed by the addition of diethylzinc and substituted hydroximoyl chlorides 46, the isoxazolidines 47 are formed with impressive enantioselectivities of up to 96% ee (Scheme 6.33) [76].
Scheme 6.33
In an extension of this work they developed a catalytic version of the reaction in which the chiral ligand (R,R)-diisopropyltartrate (DIPT) was applied in 20 mol% [77]. In spite of the reduction of the amount of the chiral ligand similar high enantioselectivities of up to 93% ee were obtained in this work. The addition of a
236 6 Asymmetric Metal-catalyzed 1,3-Dipolar Cycloaddition Reactions
small amount of 1,4-dioxane proved to be crucial for the enantioselectivity of the reaction. A proposal for the reaction mechanism was given and it is outlined in Scheme 6.34. Allyl alcohol 45, hydroximoyl chloride 46 and diethylzinc react to form 48, which is mixed with the ligand and an additional amount of diethylzinc to form 49. The achiral complex 48 is apparently much less activated for a 1,3-di- polar cycloaddition reaction compared to 49, which controls the enantioselectivity of the reaction. The increased reactivity of 49 compared to 48 might be because of a ligand-accelerating effect of DIPT when coordinated to the metal. After formation of the isoxazoline 50, the Zn-DIPT moiety of 50 proceeds in the catalytic cycle and 51 is formed. When the reaction is complete 51 is hydrolyzed to give 47 in up to 93% ee (R = t-Bu) [77].
Scheme 6.34
The above described approach was extended to include the 1,3-dipolar cycloaddition reaction of nitrones with allyl alcohol (Scheme 6.35) [78]. The zinc catalyst which is used in a stoichiometric amount is generated from allyl alcohol 45, Et2Zn, (R,R)-diisopropyltartrate (DIPT) and EtZnCl. Addition of the nitrone 52a leads to primarily trans-53a which is obtained in a moderate yield, however, with high ee of up to 95%. Application of 52b as the nitrone in the reaction leads to higher yields of 53b (47–68%), high trans selectivities and up to 93% ee. Compared to other metal-catalyzed asymmetric 1,3-dipolar cycloaddition reactions of

6.10 Palladium Catalysts for Reactions of Nitrones 237
nitrones, this reaction cannot be assigned as normal or inverse electron-demand. The reaction is controlled, not primarily by the alteration of FMO energies, but by the chelation of the substrates to the catalyst leading to a favorable entropy of the pseudo-intramolecular intermediates.
Scheme 6.35
It should also be mentioned that in connection with the investigations on MgX2- BOX catalysts, Desimoni et al. also tested a Zn(ClO4)2-BOX catalyst for the 1,3-di- polar cycloaddition of a nitrone and acryloyloxazolidinone (see Scheme 6.17). Contrary to the magnesium catalysts, this zinc catalyst was exo selective as an 27:73 exo/endo ratio was observed, and 84% ee of the exo isomer was obtained [51].
6.10
Palladium Catalysts for Reactions of Nitrones
For the activation of a substrate such as 19a via coordination of the two carbonyl oxygen atoms to the metal, one should expect that a hard Lewis acid would be more suitable, since the carbonyl oxygens are hard Lewis bases. Nevertheless, Furukawa et al. succeeded in applying the relative soft metal palladium as catalyst for the 1,3-dipolar cycloaddition reaction between 1 and 19a (Scheme 6.36) [79, 80]. They applied the dicationic Pd-BINAP 54 as the catalyst, and whereas this type of catalytic reactions is often carried out at rt or at 0 C, the reactions catalyzed by 54 required heating at 40 C in order to proceed. In most cases mixtures of endo-21 and exo-21 were obtained, however, high enantioselectivity of up to 93% were obtained for reactions of some derivatives of 1.
A transition structure model was proposed which accounts for the high selectivities obtained for some of the substrates [80]. In the structure shown in Scheme 6.37 the two phosphorus atoms of the Tol-BINAP ligand and the two car-

238 6 Asymmetric Metal-catalyzed 1,3-Dipolar Cycloaddition Reactions
Scheme 6.36
bonyl oxygens of the crotonoyl oxazolidinone are arranged in a square planar fashion around the palladium center (note that the counter-ions are omitted from this model). From the model the upper si face of the alkene is sterically available for the cycloaddition reaction, while the re face is shielded by one of the Tol-BINAP p- tolyl groups.
Scheme 6.37
Furukawa et al. also applied the above described palladium catalyst to the inverse electron-demand 1,3-dipolar cycloaddition of nitrones with vinyl ethers. However, all products obtained in this manner were racemic [81].

6.11 Lanthanide Catalysts for Reactions of Nitrones 239
6.11
Lanthanide Catalysts for Reactions of Nitrones
In 1997 the application of two different chiral ytterbium catalysts, 55 and 56 for the 1,3-dipolar cycloaddition reaction was reported almost simultaneously by two independent research groups [82, 83]. In both works it was observed that the achiral Yb(OTf)3 and Sc(OTf)3 salts catalyze the 1,3-dipolar cycloaddition between nitrones 1 and alkenoyloxazolidinones 19 with endo selectivity. In the first study 20 mol% of the Yb(OTf)2-pyridine-bisoxazoline complex 55 was applied as the catalyst for reactions of a number of derivatives of 1 and 19. The reactions led to endo-selective 1,3-dipolar cycloadditions giving products with enantioselectivities of up to 73% ee (Scheme 6.38) [82]. In the other report Kobayashi et al. described a 1,3-dipolar cycloaddition catalyzed by 20 mol% of the Yb(OTf)3–BINOL complex 56 in the presence of the achiral tertiary amine 57 [83]. In this approach the nitrone 1 was formed in situ from the respective aldehyde and hydroxylamine. High endo selectivities were observed and for one derivative the product endo-21 (R1 = Bn, R2 = Me) was obtained with 78% ee [83]. In an extension of these investigations the 1,3-dipolar cycloaddition reaction was performed in the presence of 20 mol% of the catalyst 56 and 40 mol% of a the chiral amine 58 [84]. By substituting the achiral amine 57 with the complex chiral amine 58 the selectivity of the reaction was largely improved. For the reactions of some derivatives of 1 and 19, endo-21 was obtained as a single diastereomer and enantioselectivities of up to 96% ee were achieved. Further investigation in this field by Kobayashi et al. led to the finding that the absolute stereoselectivity of the reaction was reversed when the reaction was performed in the absence of MS 4 Å [85]. This observation makes an analog to the MgX2-BOX-catalyzed reactions, where a similar incidence was observed [49]. In the reaction catalyzed by 56 using 58 as the additive endo-21 (R1 = Bn, R2 = Me) was obtained in 96% ee in the presence of MS 4 Å. In the absence of MS 4 Å the opposite enantiomer was obtained in 50% ee. This inverse selectivity could be improved by using various N-oxides as a third additive [85].
Whereas there are numerous examples of the application of the products from diastereoselective 1,3-dipolar cycloaddition reaction in synthesis [7, 8], there are only very few examples on the application of the products from metal-catalyzed asymmetric 1,3-dipolar cycloaddition reaction in the synthesis of potential target molecules. The reason for this may be due to the fact that most metal-catalyzed asymmetric 1,3-dipolar cycloaddition reaction have been carried out on model systems that have not been optimized for further derivatization. One exception of this is the synthesis of a -lactam by Kobayashi and Kawamura [84]. The isoxazolidine endo-21b, which was obtained in 96% ee from the Yb(OTf)3-BINOL-catalyzed 1,3-dipolar cycloaddition reaction, was converted into the ester derivative 59, quantitatively. Hydrogenation over palladium on carbon opens the isoxazolidine ring and cleaves the N-benzyl moiety to give 60. Following silyl protection of the hydroxy group in 60, the final ring-closure is mediated by LDA to give the -lactam 62 in a high yield with a conserved optical purity of 96% ee (Scheme 6.39).