

Cycloaddition Reactions in Organic Synthesis
.pdf98 3 Enantioselective [2+1] Cycloaddition: Cyclopropanation with Zinc Carbenoids
Fig. 3.3 1H NMR and X-ray crystallography study of the bis-ether complex 28. [Denmark, S.E.; Edwards, J.P.; Wilson, S.R. J. Am. Chem. Soc. 1992, 114, 2592. Reprinted with permission from The American Chemical Society]
the two methylene groups. The two signals could not be resolved, even at low temperatures, attesting to the rapid rate of exchange.
Most importantly, this complex afforded crystals suitable for X-ray analysis. This provides the first crystallographic data regarding a zinc carbenoid. Examination of the X-ray structure confirms that the initially formed carbenoid reagent is Zn(CH2I)2 (Fig. 3.3) [33]. The crystal structure reveals several interesting features of this complex. The complex crystallizes as a monomer, although two distinct molecules are visible within one unit cell. The differences in the two structures are minor. The two iodomethyl groups are clearly distinguished in the solid state, despite their coalescence on the NMR time-scale. The upper group is in close contact with the dimethyl bridge of the ether ligand. To avoid unfavorable steric interactions, the iodine is positioned out and away from the bis-ether backbone. The lower group, which does not encounter the steric bulk of the dimethyl bridge, is rotated in an opposite orientation leaving the iodine over the zinc chelate ring. The disposition of the two iodomethyl groups around the central zinc atom is slightly bent (C–Zn–C = 138.5 ). Although such organozinc species are assumed to be linear around the zinc atom, this deviation can easily be explained by the influence of the coordinated oxygen atoms. In terms of bond lengths, the attachment of the ether ligand seems to have little effect on the gross structural features of the carbenoid. The carbon-iodine bonds are not elongated relative to a typical sp3-carbon bound iodide (2.13–2.21 Å) [34 a]. It is also interesting to note that bond lengths of the zinc-carbon bonds are equivalent to those observed in the crystal structures of other alkylzinc species (1.92–1.98 Å) [34 b–e].
Although this study is a major advance in clarifying the structure of these organozinc reagents, it did not, at the time, address the Schlenk equilibrium issue. Because ZnI2 is the sole by product of the methylene transfer process, its concentration is constantly increasing over the course of a cyclopropanation. Thus, whereas Zn(CH2I)2 may accurately describe the preformed zinc carbenoid, disproportionation with ZnI2 formed during the reaction is a distinct possibility. Thus, it is imperative to investigate the influence of the zinc iodide, ZnI2, on the composition of the initially formed reagent. If the ZnI2 formed can combine with Zn(CH2I)2

3.3 Structure and Dynamic Behavior of Zinc Carbenoids 99
to generate the putative Simmons-Smith reagent, ICH2ZnI, then the possibility that ICH2ZnI is the active species throughout the cyclopropanation cannot be excluded.
To gain information on the interconversion of all species concerned (Zn(CH2I)2, ZnI2, and ICH2ZnI); Charette et al. made recourse to the use of Denmark’s chelating bis ether 27 to slow the exchange process and to facilitate spectroscopic identification of the species. The experimental design used by Charette et al. approached the establishment of this Schlenk equilibrium from the other side (Scheme 3.14) [35]. Formation of ICH2ZnI in the presence of the chiral bis ether 27 leads to clean formation of the complex 29, which is characterized by 1H and 13C NMR spectroscopy. This compound clearly differs from the complex 27- Zn(CH2I)2 (28) reported by Denmark et al. Formation of the complex 27-ZnI2 (30) also leads to a unique set of signals. When the solution behavior of 29 is observed over a variety of temperatures, the related complexes 28 and 30 are never observed, suggesting that ICH2ZnI is the thermodynamically favored species. Although 29 could not be crystallized, Charette has obtained a crystal structure of the corresponding complex of ICH2ZnI with 18-crown-6 31 (Fig. 3.4) [36].
This conclusion must, however, be tempered with two important caveats. Firstly the conclusion is based on a negative experiment; the lack of change of the com-
Scheme 3.14
Fig. 3.4 X-ray crystal structure of the 18-crown-6-ICH2ZnI adduct 31. [Charette, A.B.; Marcoux, J.F.; Bélanger-Gariépy, F. J. Am. Chem. Soc. 1996, 118, 6792. Reprinted with permission from The American Chemical Society]

100 3 Enantioselective [2+1] Cycloaddition: Cyclopropanation with Zinc Carbenoids
plex 29 could simply mean that ICH2ZnI is inert to disproportionation while complexed. To resolve this, the equilibrium should also be approached from the other direction by combining 28 and 30. If no equilibration occurs then unfortunately the experiment is flawed by the perturbation introduced by the need for complexing these agents. A more conclusive study on the role of the Schlenk equilibrium in course of cyclopropanation with Furukawa-type reagents was reported by Denmark and will be described in a later section on asymmetric catalysis. Thus, even though Zn(CH2I)2 is the direct product of the Furukawa preparation, its role in the cyclopropanation is probably less important than that of ICH2ZnI.
These foregoing studies have placed the chemistry of zinc carbenoids on a more solid structural footing. The early conclusion that ICH2ZnI was the active species formed under Simmons-Smith conditions was solely based upon titration studies, techniques unable to provide insight into rapid dynamic processes occurring in solution. With the advent of NMR spectroscopy, more in depth investigations lent clarity to this complex situation. By use of the Furukawa procedure, the initially formed species is clearly Zn(CH2I)2. However, in the presence of ZnI2, disproportionation to ICH2ZnI occurs. Since ZnI2 is the inorganic byproduct of the cyclopropanation, earlier studies which assumed Zn(CH2I)2 to be the active reagent would have to be re-evaluated. Not only do these carbenoid species differ in terms of structure, they also differ in terms of reactivity and selectivity. This reevaluation will be particularly important for understanding the role of ZnI2 in the asymmetric variant of the cyclopropanation.
3.4
Stereoselective Simmons-Smith Cyclopropanations
3.4.1
Substrate-directed Reactions
The landmark report by Winstein et al. (Scheme 3.6) on the powerful accelerating and directing effect of a proximal hydroxyl group would become one of the most critical in the development of the Simmons-Smith cyclopropanation reactions [11]. A clear syn directing effect is observed, implying coordination of the reagent to the alcohol before methylene transfer. This characteristic served as the basis of subsequent developments for stereocontrolled reactions with many classes of chiral allylic cycloalkenols and indirectly for chiral auxiliaries and catalysts. A full understanding of this phenomenon would not only be informative, but it would have practical applications in the rationalization of asymmetric catalytic reactions.
In 1963, Dauben and Berezin published the first systematic study of this syn directing effect (Scheme 3.15) [37]. They found that the cyclopropanation of 2-cyclohexen-1- ol 32 proceed in 63% yield to give the syn isomer 33 as the sole product. They observed the same high syn diastereoselectivity in a variety of cyclic allylic alcohols and methyl ethers. On the basis of these results, they reasonably conclude that there must be some type of coordinative interaction between the zinc carbenoid and the substrate.

3.4 Stereoselective Simmons-Smith Cyclopropanations 101
Scheme 3.15
For free hydroxyl groups the question remained if this was a simple coordinative interaction or if the reaction was proceeding through a zinc alkoxide. The sensitivity of these reagents to protic agents (water and alcohols) is well known, so that zinc alkoxide formation is a reasonable possibility. The effective directing ability of a methyl ether (vide infra) seems to substantiate simple coordination. The yields of the cyclopropanations also support the notion of coordination between the Lewis acidic zinc atom and the oxygen as shown in ii (Scheme 3.16). Since 1.2 equivalents of diiodomethane are used, yields of the cyclopropane could not exceed 20% if reaction were occurring through the zinc alkoxide iii. However, yields as high as 82% are common in this study. Hydrolysis may be possible, but it is not competitive with methylene transfer, resulting in the formation of the product-zinc iodide complex iv. Dauben and Berezin suggest that the syn directing effect is a direct result of a transition structure where the zinc reagent is simply coordinated to the hydroxyl group as shown in ii. It was proposed that coordination of the zinc reagent to an axially disposed hydroxyl is required to account for these observations. This hypothesis was tendered on the reasonable assertion that an axial hydroxyl group is in closer proximity to the double bond than an equatorial group.
Scheme 3.16
In 1968, Chan and Rickborn undertook a more detailed study of this directing effect [13]. Rather than rely on selectivity and yield as indicators of the effect of the oxygen substituent, they examined the rates of the cyclopropanations. At this point, little kinetic data was available for this reaction due to technical difficulties involved in monitoring the progress of a heterogeneous process. Simmons and Blanchard had previously determined that cyclopropanation was first order in both alkene and zinc reagent (presuming ICH2ZnI). Because the concentration of the zinc reagent is difficult to assess, absolute rates could not be determined. Rickborn reasoned that relative rates would be indicative of the influence of the oxygen substituent. In this study a variety of vinylic, allylic and homoallylic alcohols and ethers were surveyed (Scheme 3.17). It is clear that the proximity of the
102 3 Enantioselective [2+1] Cycloaddition: Cyclopropanation with Zinc Carbenoids
oxygen substituent has a strong effect on the observed rate enhancement. The greater the distance between the hydroxyl and alkene groups, the slower the reaction. This becomes clear upon comparing alcohols 32, 38, and 40. The reactivity of the enol ether 34 initially appears inconsistent with this trend, but it can be explained by the fact that it is an ether rather than a free hydroxyl group. As can be seen, the methyl ether 36 is less reactive than the corresponding alcohol 32. In almost all cases, complete syn diastereoselectivity is observed. An erosion in stereoselectivity is only observed in the case of the poorly reactive -alkenol 40.
Scheme 3.17
These results illustrate the importance of positioning an oxygen substituent proximal to the double bond for enhanced rate and selectivity. Rickborn also sought to test Dauben and Berezin’s earlier conclusions regarding the involvement of an axial hydroxyl group. Unfortunately, the results from alcohols 32 and 38 cannot differentiate the involvement of an axial or equatorial hydroxyl group. Therefore, to test this assertion, Rickborn examined the relative rates of two different substituted allylic alkenols, cis- and trans-5-methyl-2-cyclohexen-1-ols 42 and 44 (Scheme 3.18). For each of the two isomeric cyclohexenols, two limiting conformations are possible. In the cis isomer (42), the preferred conformer has both substituents in an equatorial position (42 e). In the trans isomer (44) the two conformers are more closely balanced but because of the smaller A value of the hydroxyl group compared to a methyl group (0.54 kcal mol–1 compared with 1.74 kcal mol–1), the major conformer should be that with the hydroxyl group axially oriented (44 a) [38]. If the cyclopropanation occurs through coordination to an axially disposed hydroxyl group as suggested by Dauben, then trans isomer 44 should react more quickly. Conformation 44 a of the trans isomer is free of trans-annular steric interactions which would hinder the reaction through an axial hydroxyl. In the cis isomer the conformation that

3.4 Stereoselective Simmons-Smith Cyclopropanations 103
places the hydroxyl group in an axial position is highly unfavored (42 a). Moreover, it should be expected that a zinc complex of this compound would not be reactive because of the severe steric interactions between the axial methyl group and the zinc complex. On the other hand, if reaction proceeds through coordination to an equatorial hydroxyl substituent, then the trans isomer should react more quickly because of the high population of conformer 42 e compared to 42 a.
Scheme 3.18
As is apparent from Scheme 3.19, the cis isomer 42 reacts three times as quickly as the trans isomer 44. In contrast to the assertion of Dauben and Berezin, the experiments demonstrate that coordination must be occurring through an equatorial hydroxyl group. This result has interesting implications for the mechanism of the methylene transfer. Examination of molecular models shows that it is not possible for an ICH2Zn unit to bridge the space between the alkene and such a remotely aligned directing group.
Scheme 3.19
To resolve this problem, Rickborn made an ingenious proposal that implicated the intermediacy of a bimetallic transition structure assembly v involving a bridging ZnI2 molecule (Fig. 3.5). This would accommodate the needed spatial requirements of the methylene transfer process. The importance of the polymetallic re-

104 3 Enantioselective [2+1] Cycloaddition: Cyclopropanation with Zinc Carbenoids
Fig. 3.5 Bimetallic transition state assembly for directed cyclopropanation
agent in the transition state assembly will become clear in the discussion of chirally modified reagents. It should be noted that this conclusion, although well justified, is not universal with respect to all zinc carbenoid-mediated cyclopropanations. There are some cases where Rickborn’s conclusion is overlooked in place of the simpler, monomeric zinc model. Rather than there being a single, unified picture for the transition structure assembly, the situation is substrate specific.
The strong directing effect of hydroxyl groups on the diastereoselectivity of the cyclopropanation process is general, although the outcome is not always intuitively obvious. For example, it has often been observed that in large ring cycloalkenols, there are high levels of anti diastereoselectivity. This contrasts the high cis stereoselectivity observed in 5-, 6- and 7-membered ring systems. Winstein et al. has provided a clear illustration of this selectivity switch and has rationalized it on the basis of the conformational analysis of large ring systems (Fig. 3.6) [39]. In the case of 8-membered rings, where a 99.5 to 0.5 anti dr is observed (46), one must carefully consider the possible conformations of a cyclooctene. The dihedral angle (C1–C2–C3–C4) in cis-cyclooctene is known to be 90 o (Fig. 3.6) [40]. In such a conformation as vi, the dihedral angle between the alkene and the hydroxyl becomes 150 o (C1–C2–C3–OH)). Examination of the full structure shows the disposition of the hydroxyl group relative to the anti face of the alkene. Again, careful conformational analysis is able to accurately predict the selectivity of Sim- mons-Smith cyclopropanations. The mechanistic rationale for the stereoselectivity of the directed cyclopropanation process is maintained. It is interesting to note that similar anti diastereoselectivity is also observed in the epoxidation of cyclooctenes with peracids [41].
Fig. 3.6 Conformational analysis of the 2-cycloocten-1-ol 46

3.4 Stereoselective Simmons-Smith Cyclopropanations 105
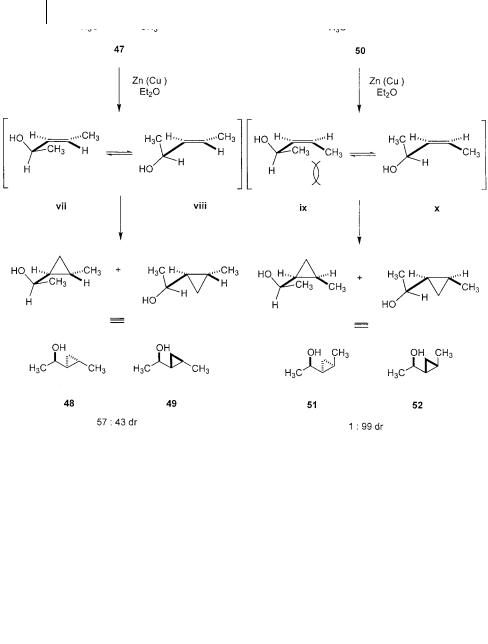
High diastereoselectivity is also observed in the cyclopropanation of acyclic allylic alcohols. Despite the conformational flexibility of these systems, they often exhibit high selectivity based on the minimization of allylic strain. Such effects are common to the chemistry of oxygen-directed epoxidations [42], hydroborations [43], dihydroxylations [44], and hydrogenations [45]. So it is not surprising that similar stereoselectivity will be manifest in the Simmons-Smith cyclopropanation, a reaction which is known to be highly dependent on a coordinative interaction. Examination of the reaction of two isomeric 3-penten-2-ols 47 and 50 shows high diastereoselectivity which is strongly dependent on the double bond configuration (Scheme 3.20) [46]. Even though this study was preformed using a racemic mixture of alcohols, information on the relative stereochemistry is still important. The stereochemical imperative of a 150 o torsional angle between the hydroxyl group and the alkene plane has important consequences for each of the reactive
conformations (vii–x). In the case of the E-substituted alkenol 47, the inside substituent, either methyl group in vii or the H in viii avoids serious A(1,3) strain by
its disposition 30 o above the plane of the alkene. Consequently, poor diastereoselectivity is observed due to the small difference in destabilizing interactions between the two competing transition states. However, the Z-substituted alkenol 50 leads to formation of the syn diastereomer 52 with high selectivity. The inside group at the allylic position again is lifted 30 o above the plane of the alkene. In this
case though, this twist is not able to alleviate the severe steric interactions between the two methyl groups in ix. The difference in A(1,3) strain between ix and x is much
larger than in 47. The conformer x is able to accommodate both the requisite disposition of the hydroxyl group and also avoid serious non-bonded interactions.
Recently, Charette et al. have also demonstrated this behavior in the stereoselective cyclopropanations of a number of enantiopure acyclic allylic ethers [47]. The high degree of acyclic stereocontrol in the Simmons-Smith cyclopropanation has been extended to synthesis several times, most notably in the synthesis of small biomolecules. Schöllkopf et al. utilized this method in their syntheses of cyclopro- pane-containing amino acids [48 a, b]. The synthesis of a cyclopropane-containing nucleoside was also preformed using acyclic stereocontrol [48 c].
Another interesting example of acyclic stereocontrol with an allylic alcohol is seen in the cyclopropanation of -hydroxysulfoximines (Scheme 3.20). The -hy- droxysulfoximine functionality is a useful directing group in a number of diastereoselective reactions [49]. In the synthesis of (–)-rothrockene 53, Johnson et al. attempted the directed Simmons-Smith cyclopropanation of the E isomer of the dienyl -hydroxysulfoximine 54 [50 a]. As shown in the previous study on acyclic allylic
alcohols, such E configured substrates typically give poor diastereoselectivities due to the absence of A(1,3) strain. However, in this case, formation of the desired product
55 is exclusive. Clearly, the presence of the neighboring sulfoximine exerts a powerful effect on the process even though coordination of the reagents is most likely occurring through the proximal hydroxyl moiety. A -hydroxysulfoximine has also been used successfully in the synthesis of tricyclic sesquiterpene thujopsene [50b].
Although the previous example involving a -hydroxysulfoximine still relies on the involvement of an alcohol, hydroxyl groups are not unique in their ability to
106 3 Enantioselective [2+1] Cycloaddition: Cyclopropanation with Zinc Carbenoids
||| |
||| |
|
Scheme 3.20
Scheme 3.21
direct the approach of the carbenoid to the alkene. Esters can also serve as directing groups for the cyclopropanation reaction. In the case of the dihydropyrrole 56, a modest degree of syn selectivity is observed (Scheme 3.22) [51]. This sense of diastereoselection is as expected in such a rigid, conformationally biased system.

3.4 Stereoselective Simmons-Smith Cyclopropanations 107
Scheme 3.22
Coordination of the carbenoid reagent to nitrogen atoms provides a productive interaction as well. Secondary acetamides have been shown to exert a strong directing affect on Simmons-Smith cyclopropanations. A moderate degree of syn direction is observed in the cyclopropanation of 1-trifluoroacetamido-2-cyclohexene 59 (Scheme 3.23, Eq. 1) [52]. Formation of the tertiary amide via methylation (61) leads to a lessening of the directing effect as evidenced by the decreased isolated yield of the syn product 62. Further studies demonstrate that coordination to an acetamido group may be favored over a hydroxyl group [53]. The cyclopropanation of the disubstituted cyclopentene 63 proceeds with exclusive syn diastereoselectivity relative to the acetamide functionality (Scheme 3.23, Eq. 2). Selectivity can be reversed by benzoylation of the acetamide (66), re-establishing the directing effect of the alcohol by blocking the coordination site on the amide nitrogen. Despite the compelling nature of this effect, questions regarding the degree of the change in selectivity must be raised due to the low yields observed in the cyclopropanation process.
Scheme 3.23
The discovery of viable substrate-direction represents a major turning point in the development of the Simmons-Smith cyclopropanation. This important phenomenon underlies all of the asymmetric variants developed for the cyclopropanation. However, more information regarding the consequences of this coordinative interaction would be required before the appearance of a catalytic, asymmetric method. The first steps in this direction are found in studies of chiral auxiliary-based methods.