

Cycloaddition Reactions in Organic Synthesis
.pdf8.4 1,3-Dipolar Cycloaddition Reactions of Nitrones 321
energy by 5.0–6.7 kcal mol–1 compared to the uncatalyzed reaction. The transitionstate energies for the reaction of nitroethene with propene was reduced from 20.7 to 13.8 kcal mol–1 by coordination of BF3 to the oxygen atom of nitroethene. The lower transition-state energy was explained in terms of a stronger interaction between the LUMOdiene and HOMOdienophile, as the LUMO energy of the BH3-coordinated nitroethene was 0.78 eV lower compared to nitroethene (–2.60 eV). The BH3 catalyst enhances the asynchronicity of the transition state due to an increase of the oxygencarbon distance. However, the most notable effect of the Lewis acid as catalyst was the delocalization of the negative charge that being transferred in the transition state; the BH3 fragment accepts 0.27 electron of the 0.31 electron transferred to the heterodiene system. It was stated that the role of the Lewis acid was to increase the electrophilic character of the nitroethene due to a stabilization of the corresponding transition-state structure through a delocalization of the negative charge that is being transferred along the nucleophilic attack of the substituted ethene [29].
The number of theoretical investigations of hetero-Diels-Alder reaction is very limited. The few papers dealing with this class of reactions have shown that the influence of the Lewis acid on the reaction course can to a high extent be compared to those found the carbo-Diels-Alder reactions. At the present stage of investigations, however, more work is needed if we are to understand the influence and control of selectivity in Lewis acid-catalyzed hetero-Diels-Alder reaction – we are probably at the beginning of a new era in this field.
8.4
1,3-Dipolar Cycloaddition Reactions of Nitrones
The 1,3-dipolar cycloaddition reaction of nitrones with alkenes gives isoxazolidines is a fundamental reaction in organic chemistry and the available literature on this topic of organic chemistry is vast. In this reaction until three contiguous asymmetric centers can be formed in the isoxazolidine 17 as outlined for the reaction between a nitrone and an 1,2-disubstituted alkene. The relative stereochemistry at C-4 and C-5 is always controlled by the geometric relationship of the substituents on the alkene (Scheme 8.6).
8.4.1
Frontier-orbital Interactions for 1,3-Dipolar Cycloaddition Reactions of Nitrones
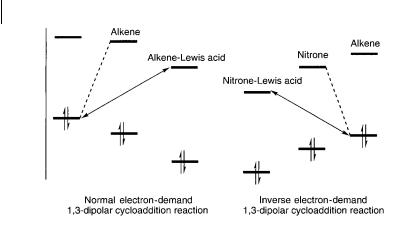
The relative FMO energies of the substrates of the 1,3-dipolar cycloaddition reaction of nitrones are important for catalytic control of the reaction. For the normal electron-demand 1,3-dipolar cycloaddition reactions the dominant FMO interac-
Scheme 8.6
322 8 Theoretical Calculations of Metal-catalyzed Cycloaddition Reactions
Fig. 8.17 An FMO diagram of the normal |
cloaddition reactions of a nitrone with an alkene, |
and inverse electron-demand 1,3-dipolar cy- |
in the absence and the presence of a Lewis acid |
tion is that of the HOMOnitrone with the LUMOalkene as outlined to the left in Fig. 8.17. The inverse electron-demand 1,3-dipolar cycloaddition reactions are dominated by the interaction between the LUMOnitrone and the HOMOalkene. By the application of a Lewis acid, the LUMO energy of the alkene is lowered by coordination of, e.g., the , -unsaturated carbonyl to the Lewis acid. As a result of the decreased energy gap between the interacting FMOs a rate acceleration of the reaction is achieved as shown to the left in Fig. 8.16. While for the inverse elec- tron-demand 1,3-dipolar cycloaddition reaction of nitrones with electron-rich alkenes the application of Lewis acids will lower the LUMO energy of the nitrone and thus a more feasible interaction with the alkene becomes possible as schematically outlined to the right in Fig. 8.17 [30].
One of the problems related to the Lewis acid activation of , -unsaturated carbonyl compounds for the reaction with a nitrone is the competitive coordination of the nitrone and the , -unsaturated carbonyl compound to the Lewis acid [30]. Calculations have shown that coordination of the nitrone to the Lewis acid can be more feasible than a monodentate coordination of a carbonyl compound. However, this problem could be circumvented by the application of alkenes which allow a bidentate coordination to the Lewis acid which is favored over the monodentate coordination.
8.4.2
Normal Electron-demand Reactions
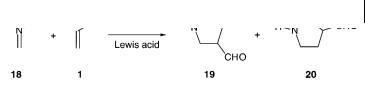
The Lewis acid-catalyzed 1,3-dipolar cycloaddition reaction of nitrones to , -un- saturated carbonyl compound in the presence of Lewis acids has been investigated by Tanaka et al. [31]. Ab-initio calculations were performed in a model reaction of the simple nitrone 18 reacting with acrolein 1 to give the two cycloadducts 19 and 20 (Scheme 8.7).
8.4 1,3-Dipolar Cycloaddition Reactions of Nitrones 323
Scheme 8.7
The uncatalyzed reaction was calculated to give isoxazolidine 19 as the preferred regioisomer with a transition-state energy of 25.7 kcal mol–1 for the formation of the exo diastereomer [31]. Two types of catalytic reactions were investigated; first acrolein 1 was activated by coordination to BH3, followed by coordination of the nitrone 18 to BH3. The change in energy for the reaction of these two reactions are shown in Fig. 8.18 and compared with uncatalyzed reaction. The coordination of the Lewis acid to 1 and 18, respectively, lower the energy of the reactants by 8.2 and 19.3 kcal mol–1, with the nitrone-BH3-acrolein system as the most stable combination. The calculated energies show that the 1,3-dipolar cycloaddition is most feasible by coordination of the Lewis acid to acrolein as the transition-state energy for this reaction is calculated to be 16.7 kcal mol–1, compared to 30.4 kcal mol–1 for the reaction starting with the nitrone coordinated to BH3. In the latter case, which in principle is a 1,3-dipolar cycloaddition reaction with inverse electron-demand (vide infra), the reaction is rather deactivated compared to the uncatalyzed 1,3-dipolar cycloaddition reaction. The most favorable transition-state structure shows that the coordination of the carbonyl oxygen atom of acrolein to BH3 makes the carbonyl oxygen atom more electron-withdrawing so that the -carbon atom becomes more electrophilic. This electronic change of acrolein is reflected in the transition-state structure of the reaction as the O–C bond is reduced from 1.85 Å in the uncatalyzed reaction to 1.59 Å in the Lewis acid-catalyzed reaction, while the C–C bond is increased from 2.35 Å to 2.57 Å. The use of more a stronger Lewis acid (BF3) as the catalyst for the reaction shows that the 1,3-dipolar cycloaddition reaction leads to a step-wise reaction path taking place via, initially, the formation a Michael-adduct complex intermediate, followed by the ring-closure step. In the Michael-adduct complex the O–N and C–C bond lengths are calculated to be 3.04 Å and 1.53 Å, respectively; i.e. the stronger Lewis acid polarize the , -unsaturated carbonyl compound further which leads to the change in reaction path in the initial phase of the reaction.
8.4.3
Inverse Electron-demand Reactions
The other catalytic approach to the 1,3-dipolar cycloaddition reaction is the inverse electron-demand (Fig. 8.17, right), in which the nitrone is coordinated to the Lewis acid, which for the reaction in Scheme 8.7 was found to be deactivated compared to the uncatalyzed reaction. In order for a 1,3-dipolar cycloaddition to proceed under these restrictions the alkene should be substituted with electron-donat- ing substituents.

324 8 Theoretical Calculations of Metal-catalyzed Cycloaddition Reactions
Fig. 8.18 Energy profile for the 1,3-dipolar cycloaddition reaction of the nitrone 18 with acrolein 1 under uncatalyzed reaction conditions, and in the presence of BH3 as the Le-
wis acids. Energies (kcal mol–1) are relative to the total energy of the starting compounds and bond lengths are in Å [31]
This reaction has been investigated by Domingo [32] using DFT calculations for the reaction of the dimethyl nitrone 21 with ethyl vinyl ether 22 (Scheme 8.8). The uncatalyzed reaction was calculated to proceed with formation of the ortho

8.4 1,3-Dipolar Cycloaddition Reactions of Nitrones 325
compounds 23, because of the lowest activation energy, ca. 14 kcal mol–1 (depending on the exo/endo selectivity) and being the most exothermic reaction of ca. 27 kcal mol–1, which is ca. 8 kcal mol–1 and ca. 6–9 kcal mol–1 lower in energy compared to the reaction path leading to the meta compounds 24. The difference in activation enthalpy for the formation of endo-23 and exo-23 is < 1 kcal mol–1 with the lowest activation for the exo stereoselection, which is opposite to the calculations by Houk et al. [33] who found a 0.8 kcal mol–1 difference for the two transition states. It was pointed out by Domingo [32] that the relative low energy difference for the transition states agrees with the fact that the endo/exo selectivity for these 1,3-dipolar cycloaddition reactions depends on the bulk of the substituents present on both the nitrone and the substituted alkene.
Scheme 8.8
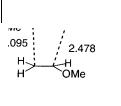
The Lewis acid-catalyzed reaction of nitrone 21 with ethyl vinyl ether 22 (Scheme 8.8) was also investigated for BH3 and AlMe3 coordinated to 21 [32]. The presence of BH3 decreases the activation energy for the formation of 23 by 3.1 and 4.5 kcal mol–1 to 9.6 kcal mol–1 for the exo-selective reaction and 11.6 kcal - mol–1 for the endo-selective reaction, respectively, while the activation energy for the formation of 24 increases by > 1.4 kcal mol–1, compared to those for the uncatalyzed reaction. The transition-state structure for the BH3-exo-selective 1,3-dipolar cycloaddition reaction of nitrone 21 with ethyl vinyl ether 22 is shown in Fig. 8.19.
The influence of the Lewis acid catalyst can be understood from the FMO diagram to the right in Fig. 8.17. The Lewis acid catalyst enhances significantly the asynchronicity of the bond-forming process for the more favorable ortho transition state as the O–C distance in the BH3-catalyzed reaction is 2.478 Å compared to 2.284 Å in the uncatalyzed reaction. For the use of AlMe3 as the catalyst the O–C distance is calculated to be 2.581 Å in the transition state.
The role of the Lewis acid in these inverse electron-demand 1,3-dipolar cycloaddition reactions can be accounted for by an increase in the electrophilic character of the nitrone by the coordination to the Lewis acid. This gives a stabilization of the corresponding transition state by delocalization of the negative charge that is being transferred along the asynchronous cycloaddition process. The role of the Lewis acid on the regioselectivity the ortho selectivity is favored by charge transfer
326 8 Theoretical Calculations of Metal-catalyzed Cycloaddition Reactions
Fig. 8.19 The calculated transition-state structure for the BH3- exo-selective 1,3-dipolar cycloaddition reaction of nitrone 21 with ethyl vinyl ether 22 [32]
by the presence of the nitrone oxygen atom, which stabilizes the incipient positive charge that is developed on the carbon atom of the substituted alkene during the cycloaddition process.
The theoretical investigations of Lewis acid-catalyzed 1,3-dipolar cycloaddition reactions are also very limited and only papers dealing with cycloaddition reactions of nitrones with alkenes have been investigated. The Influence of the Lewis acid catalyst on these reactions are very similar to what has been calculated for the carboand hetero-Diels-Alder reactions. The FMOs are perturbed by the coordination of the substrate to the Lewis acid giving a more favorable reaction with a lower transition-state energy. Furthermore, a more asynchronous transition-struc- ture for the cycloaddition step, compared to the uncatalyzed reaction, has also been found for this class of reactions.
8.5
Summary
The investigation of the metal-catalyzed cycloaddition reactions is in its beginning. We now begin to have the computational methods necessary for doing reliable calculations on systems being used in the laboratory. This chapter has shown that the one now begin to understand cycloaddition reaction from a theoretical point of view. There is, however, still a long way to go before computational chemists begin to understand and predict catalytic enantioselective cycloaddition reactions.
Acknowledgments
This work was made possible by a grant from The Danish National Research Foundation.
References
[1] |
See, e.g. (a) Woodward, R. B.; Hoff- |
[3] |
See, e.g. (a) Lutz, E. F.; Bailey, G. M. J. |
|
mann, R.; The Conservation of Orbital |
|
Am. Chem. Soc. 1964, 86, 3899; (b) Inu- |
|
Symmetry; Verlag Chemie 1970; (b) Flem- |
|
kai, T.; Kojima, T. J. Org. Chem. 1966, |
|
ing, L. Frontier Orbitals and Organic |
|
31, 1121; 1970 35, 1342; 1971, 36, 924. |
|
Chemical Reactions, John Wiley and Sons, |
[4] |
See, e.g. (a) Inukai, T.; Kojima, T. J. Org. |
|
London, 1977. |
|
Chem. 1967, 32, 869, 872, 2032; (b) |
[2] |
Houk, K. N.; Strozier, R. W. J. Am. |
|
Sauer, J.; Kredel, J. Tetrahedron Lett. |
|
Chem. Soc. 1973, 95, 4094. |
|
1966, 731. |
|
|
|
References |
327 |
|
|
|
|
|
[5] |
Loncharich, R. T.; Brown, F. K.; Houk, |
[20] |
Garcia, J. I.; Martínez-Marino, V. J. |
|
|
K. N.; J. Org. Chem. 1989, 54, 1129. |
|
Org. Chem. 1998, 63, 2321. |
|
[6] |
Birney, D. M.; Houk, K. N.; J. Am. |
[21] |
Sabi, A.; Branchadell, V.; Artuno, |
|
|
Chem. Soc. 1990, 112, 4127. |
|
R. M.; Oliva, A. J. Org. Chem. 1997, 62, |
|
[7] |
Torri, J.; Azzaro, M. Bull. Soc. Chim. Fr. |
|
3049. |
|
|
1978, II-283. |
[22] |
Avalos, M.; Babiano, R.; Bravo, J. L.; |
|
[8] |
See, e.g. (a) Loncharich, R. J.; |
|
Cintas, P.; Jiménez, J. L.; Palacios, J. C.; |
|
|
Schwarts, T. R.; Houk, K. N.;, J. Am. |
|
Silva, M. A. J. Org. Chem. 2000, 65, 6613. |
|
|
Chem. Soc. 1987, 109, 14; (b) Güner, |
[23] |
Ishihara, K.; Kondo, S.; Kurihara, H.; |
|
|
O. F.; Ottenbrite, R. M.; Shillady, |
|
Yamamoto, H. J. Org. Chem. 1997, 62, |
|
|
D. D.; Alson, P. V. J. Org. Chem. 1987, |
|
3026. |
|
|
52, 391; (c) LePage, T. J.; Wiberg, K. B. J. |
[24] |
See, e.g. (a) de la Torre, M. F.; Caballo, |
|
|
Am. Chem. Soc. 1988, 110, 6642. |
|
M. C.; Whiting, A. Tetrahedron 1999, 55, |
|
[9] |
Yamabe, S.; Dai, T.; Minato, T. J. Am. |
|
8547; (b) Hunt, I. R.; Rauk, A.; Keay, |
|
|
Chem. Soc. 1995, 117, 10994. |
|
B. R. J. Org. Chem. 1996, 61, 751; (c) |
|
[10] |
(a) Garcia, J. I.; Mayoral, J. A.; Salvatel- |
|
Domingo, L. R.; Picher, M. T.; Andrés, |
|
|
la, L. J. Am. Chem. Soc. 1996, 118, |
|
J. J. Phys. Org. Chem. 1999, 12, 24; (d) |
|
|
11680; (b) Garcia, J. I.; Martínez-Mari- |
|
Branchadell, V.; Oliva, A.; Bertran, J. |
|
|
no, V.; Mayoral, J. A.; Salvatella, L. J. |
|
J. Mol. Struc. (Theochem.) 1986, 31, 117; |
|
|
Am. Chem. Soc. 1998, 120, 2415. |
|
(e) Jurasic, B. S.; Zdravkovski, Z. Tetra- |
|
[11] |
Singleton, D. A. J. Am. Chem. Soc. |
|
hedron 1994, 54, 379. |
|
|
1992, 114, 6563. |
[25] |
See, e.g. (a) Tietze, L. F.; Fennen, J.; An- |
|
[12] |
Yamabe, S.; Minato, T. J. Org. Chem. |
|
ders, E. Angew. Chem. 1989, 101, 1420; |
|
|
2000, 65, 1830 |
|
(b) McCarrick, M. A.; Wu, Y.-D.; Houk, |
|
[13] |
Dai, W.-M.; Lau, C. W.; Chung, S. H.; |
|
K. N.; J. Am. Chem. Soc. 1992, 114, 1499; |
|
|
Wu, Y.-D. J. Org. Chem. 1995, 60, 8128. |
|
J. Org. Chem. 1993, 58, 3330; (c) Jursic, |
|
[14] |
For a recent review of the use of the |
|
B. S.; Zdravkovski, Z. J. Phys. Org. |
|
|
TADDOL-ligand in chemistry see See- |
|
Chem. 1994, 7, 641; (d) Tietze, L. F.; |
|
|
bach, D.; Buck, A. K.; Heckel, A. Angew. |
|
Schuffenhauser, A.; Achreiner, P. R. |
|
|
Chem. Int. Ed. 2001, 40, 92. |
|
J. Am. Chem. Soc. 1999, 120, 7952. |
|
[15] |
See, e.g. (a) Narasaka, K.; Inoue, M.; |
[26] |
Danishefsky, S. J.; Myles, D. C.; Harrey, |
|
|
Okada, N. Chem. Lett. 1986, 1109; (b) |
|
D. F. J. Am. Chem. Soc. 1987, 109, 862. |
|
|
Narasaka, K.; Inoue, M.; Yamada, T. |
[27] |
Roberson, M. I.; Jepsen, A. S.; Jørgen- |
|
|
Chem. Lett. 1986, 1967; (c) Narasaka, K.; |
|
sen, K. A.; Tetrahedron 2001, 57, 907. |
|
|
Iwasawa, N.; Inoue, M.; Nakashima, |
[28] |
Venturini, A.; Joglar, J.; Fustero, S.; |
|
|
M.; Sugimoto, L. J. Am. Chem. Soc. |
|
Gonzales, J. J. Org. Chem. 1997, 62, |
|
|
1989, 111, 5340. |
|
3919. |
|
[16] |
Gothelf, K. V.; Hazell, R. G.; |
[29] |
Domingo, L. R.; Arnó, M.; Andrés, J. J. |
|
|
Jørgensen, K. A. J. Am. Chem. Soc. 1995, |
|
Org. Chem. 1999, 64, 5867. |
|
|
117, 4435. |
[30] |
(a) Gothelf, K. V.; Jørgensen, K. A.; |
|
[17] |
(a) Gothelf, K. V.; Jørgensen, K. A.; J. |
|
Chem. Rev. 1998, 98, 863; (b) Chem. Com- |
|
|
Org. Chem. 1995, 60, 6487; (b) Gothelf, |
|
mun. 2000, 1449. |
|
|
K. V.; Jørgensen, K. A.; J. Chem. Soc., |
[31] |
Tanaka, J.; Kanemasa, S. Tetrahedron |
|
|
Perkin Trans. 2 1997, 111; (c) Gothelf, |
|
2001, 57, 899. |
|
|
K. V.; Thomsen, I.; Jørgensen, K. A.; J. |
[32] |
Domingo, L. R. Eur. J. Org. Chem. 2000, |
|
|
Am. Chem. Soc. 1996, 118, 59. |
|
2265. |
|
[18] |
Seebach, D.; Dahinden, R.; Marti, |
[33] |
Liu, J.; Niwayama, S.; You, Y.; Houk, |
|
|
R. E.; Beck, A. K.; Plattner, D. A.; Küh- |
|
K. N.; J. Org. Chem. 1998, 63, 1064. |
|
|
ler, F. N. M. J. Org. Chem. 1995, 60, |
|
|
|
|
1788. |
|
|
|
[19]Haase, C.; Sarko, C. R.; DiMare, M. J. Org. Chem. 1995, 60, 1777.