

Cycloaddition Reactions in Organic Synthesis
.pdf108 3 Enantioselective [2+1] Cycloaddition: Cyclopropanation with Zinc Carbenoids
3.4.2
Auxiliary-directed Reactions
3.4.2.1 Chiral Ketals
The powerful influence of an oxygen substituent on the rate and stereoselectivity of cyclopropanation augured well for the development of a chiral auxiliary based approach to asymmetric synthesis [54]. The design of the chiral auxiliary would take into account:
(i)the need to provide a suitable functionality for coordination of the zinc species;
(ii)the means, through a reversible protocol, to attach the auxiliary to the substrate; and
(iii)an effective, asymmetric environment ideally derived from commercially available chiral materials.
Upon removal of the auxiliary, an enantioenriched product could be obtained. The application of chiral auxiliary-based methods to Simmons-Smith cyclopropanation not only provided a useful synthetic strategy, but it also served to substantiate earlier mechanistic hypotheses regarding the directing influence of oxygen-contain- ing functional groups on the zinc reagent [6 d].
The use of chiral auxiliaries allows for extension of the Simmons-Smith method to structures which otherwise could not be generated stereoselectively in a single step. In 1990, Mash et al. reported a study of the cyclopropanation of 2-cyclohexe- nones which were modified by the formation of chiral ketals [55]. In order to access such -cyclopropyl ketones in a selective manner, a strategy is required to differentiate the enantiotopic faces of the starting enone (Scheme 3.24). The ketal functionality is well suited for this endeavor. Not only does it break the symmetry of the enone moiety through the use of a chiral diol, it provides a site for coordination of the zinc carbenoid. In the absence of the dioxolane oxygens, there is no available site for coordination and thus activation of the carbenoid. Although this strategy may seem more complex than the substrate-directed processes discussed in the foregoing section, it will become clear that similar, stepwise conformational analysis will be able to predict reactivity and selectivity in these important systems.
The initial investigation focused on the use of threitol-derived auxiliaries with various substituent groups on the dioxolane ring (Table 3.3). However, it became evident that the oxygen atoms in the substituents had a detrimental effect on selectivity. Comparison of the diastereoselectivities for the ketals 69–71, which contain Lewis basic sites in the substituents at the 1 and 2 positions, with those from simpler diol derived ketals 72–74 demonstrates the conflicting effects of numerous coordination sites. The simpler, diol-derived ketals provide superior results compared to the threitol derived ketals. The highest diastereoselectivity is observed in the case of the 1,2-diphenylethane-1,2-diol derived ketal 74.
This unexpected outcome clearly implicated an important stereodirecting role for the dioxolane oxygens. To clarify the significance of having oxygen atoms bound to the stereogenic centers, Mash carried out an important control experiment by exam-

3.4 Stereoselective Simmons-Smith Cyclopropanations 109
Scheme 3.24
ination of an all-carbon analog of acetal 74, 2,3-diphenylspiro[4.5]dec-6-ene 75 (Scheme 3.25). The lack of diastereoselectivity in the cyclopropanation of this compound clearly demonstrates the importance of the ketal oxygens.
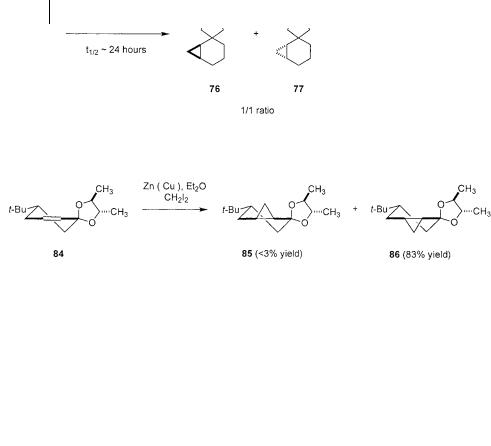
These remarkable observations stimulated an investigation to understand the origin of the directing effect. To clarify the contribution of the axiallyand equato- rially-oriented oxygen atoms in the ketal, a survey of the reaction of three conformationally biased t-butyl cyclohexenone ketals 78, 81 and 84 was undertaken (Scheme 3.26) [56]. In each case, careful conformational analysis provides critical clues to rationalizing selectivity.
In the conformationally biased cyclohexene ring, the two dioxolane oxygens must assume fixed axial and equatorial positions. Coordination of the zinc carb-
Tab. 3.3 Cyclopropanation of cyclohexenone ketals 69–74
Entry |
Ketal |
R |
Yield (%) |
dr (A : B) |
|
|
|
|
|
|
|
1 |
69 |
CH2OCH2Ph |
86 |
5 |
: 1 |
2 |
70 |
CO2CH3 |
37 |
3 |
: 2 |
3 |
71 |
CH2OH |
50 |
1 |
: 2 |
4 |
72 |
CH3 |
86 |
9 |
: 1 |
5 |
73 |
CH2CH2CH2Ph |
92 |
>9 : 1 |
|
6 |
74 |
Ph |
90 |
19 |
: 1 |
|
|
|
|
|
|
110 3 Enantioselective [2+1] Cycloaddition: Cyclopropanation with Zinc Carbenoids
Scheme 3.25
Scheme 3.26
enoid to the axial oxygen requires reaction via a transition structure involving a single zinc unit, as proposed by Dauben and Berezin. Coordination of the zinc reagent to the equatorial oxygen requires reaction via a bimetallic zinc unit, much like that proposed by Rickborn. By adopting the conclusions of Rickborn’s studies, reaction via the equatorial pathway was assumed. Although this bias is important in all three cases, the reaction of the achiral ketal will give a quantitative value for the selectivity between these two pathways.
Each oxygen atom in the dioxolane has two lone pairs available for coordination to the zinc reagent. In each case, one is proximal to the double bond and one is distal, the latter being too far away to be involved in any kind of productive interaction. The proximal lone pair of the equatorial oxygen substituent is referred to as the “topographically preferred site” (Fig. 3.7).
Further examination of the chiral ketals reveals that the lone pairs available for reagent coordination are oriented either in a syn or an anti relationship to the neighboring methyl substituents. The influence of the chiral auxiliary over the reaction is now clear. If zinc coordination must occur proximal to the double bond,

3.4 Stereoselective Simmons-Smith Cyclopropanations 111
Fig. 3.7 Stereoelectronic rationale for reagent coordination
this coordination will be influenced by the steric demands imposed by the neighboring substituent. The lone pair with the anti relationship to the ketal substituent is then the “sterically preferred site”. In terms of rationalizing the selectivity for the chiral ketals, topographic and steric effects both contribute to selectivity. In the case of diastereoisomer 81, conformational analysis shows that these effects are in conflict whereas in isomer 84, they are cooperative. Selectivity should then be higher in the latter, matched case.
The results for the cyclopropanation of the three ketals confirm these predictions (Scheme 3.26). The unsubstituted ketal 78 shows the inherent selectivity of reaction between the axial and equatorial pathways. Favored reaction via the equatorial pathway is in clear agreement with Rickborn’s earlier predictions regarding the reactive conformation. Examination of the two chiral ketals 81 and 84 confirms the prediction of the foregoing conformational analysis. The mismatched case 81 shows a low level of selectivity and reactivity. In the case where the topographic and steric biases are matched (84) the selectivity is high. These results stand as an important step in the understanding of the Simmons-Smith cyclopropanation. The ability of conformational analysis to successfully predict the relative reactivity and absolute sense of selectivity for cyclopropanation of these two chiral ketal substrates is in large measure a vindication of the insights provided by earlier mechanistic studies.
3.4.2.2 Chiral Vinyl Ethers
Another auxiliary-directed cyclopropanation reaction which provides insights into the nature of the Simmons-Smith process was reported by Tai et al. in 1988 [57]. Chiral vinyl ethers bearing a C2 symmetric pentanediol auxiliary were found to afford the corresponding cyclopropanes with high diastereoselectivity (Table 3.4). To optimize selectivity, methods for the generation of the carbenoid, as well as solvent and auxiliary structure were investigated. To generate the zinc carbenoid, the authors employed either the original Simmons-Smith procedure (CH2I2–Zn–Cu) or a modified Furukawa procedure (5 : 10 equiv. ZnEt2/CH2I2). The first substrate to be investigated was the 2,4-pentanediol modified enol ether 89. By use of the

112 3 Enantioselective [2+1] Cycloaddition: Cyclopropanation with Zinc Carbenoids
original Simmons-Smith procedure, a number of ethereal solvents were tested and, in each case, the selectivity is high. A mixture of diethyl ether and dimethoxyethane proves to be best (Table 3.4, entries 1–3). However the high temperatures required for the reaction, the modest yields and the difficulties involved in further diastereomeric enrichment of the product via recrystallization demanded a more selective method be found.
Use of the Furukawa procedure in conjunction with the 2,4-pentanediol-modi- fied enol ether 89 provided more flexibility with regard to the reaction conditions (Table 3.4, entries 4–10). After surveying a number of solvents, diethyl ether, THF and dimethoxyethane were found to be most efficient at affording high selectivities. It is, however, observed that addition of ZnI2 to the reaction mixture can also raise selectivity (Table 3.4, entries 6 and 8). It is intriguing that this additive is not observed to have an effect on rate or yield. In these reactions, a large excess of the carbenoid reagent is employed (5:10 equiv. ZnEt2/CH2I2). Although these conditions still adhere to proportions of the original Furukawa protocol, the use of excess reagent is justified by the gradual decomposition of the zinc carbenoid over the course of the reaction. This is most likely an effect of temperature; typically, the Furukawa conditions employs reduced temperatures.
A simpler solution to obtaining high selectivity is found through use of an enol ether containing a bulkier chiral auxiliary, 2,6-dimethyl-3,5-heptanediol 92 (Table 3.5). Again, a number of conditions were surveyed in order to optimize the reaction. The zinc-copper couple gives high selectivity, but it cannot compare to the exceptional selectivity obtained using the Furukawa reagent. Although selectivity
Tab. 3.4 Cyclopropanation of the chiral enol ether 89 under Simmons-Smith conditions
Entry |
Reagent |
Solvent |
ZnI2 (mol%) |
Temp. ( C) Yield (%) |
dr (90 : 91) |
||
|
|
|
|
|
|
|
|
1 |
Zn(Cu) |
THF |
0 |
60 |
65 |
90 |
: 10 |
2 |
Zn(Cu) |
Et2O |
0 |
50 |
53 |
94 |
: 6 |
3 |
Zn(Cu) |
Et2O + DME |
0 |
50 |
69 |
95 |
: 5 |
4 |
Zn(CH2I)2 |
Hexane |
0 |
0 |
52 |
70 |
: 30 |
5 |
Zn(CH2I)2 |
CH2Cl2 |
0 |
0 |
73 |
86 |
: 14 |
6 |
Zn(CH2I)2 |
CH2Cl2 |
100 |
0 |
15 |
97 |
: 3 |
7 |
Zn(CH2I)2 |
Et2O |
0 |
0 |
73 |
46 |
: 54 |
8 |
Zn(CH2I)2 |
Et2O |
100 |
0 |
65 |
84 |
: 16 |
9 |
Zn(CH2I)2 |
THF |
0 |
20 |
65 |
97 |
: 3 |
10 |
Zn(CH2I)2 |
DME |
0 |
20 |
56 |
98 |
: 2 |
|
|
|
|
|
|
|
|

3.4 Stereoselective Simmons-Smith Cyclopropanations 113
Tab. 3.5 Cyclopropanation of the chiral enol ethers 92–95 under Furukawa conditions
Entry |
Substrate |
n |
Yield (%) |
dr |
|
|
|
|
|
1 |
92 |
5 |
81 |
> 99 : 1 |
2 |
93 |
6 |
87 |
> 99 : 1 |
3 |
94 |
7 |
77 |
> 99 : 1 |
4 |
95 |
8 |
58 |
> 99 : 1 |
|
|
|
|
|
is high in a number of solvents, the reaction in diethyl ether at 20 C is found to be superior in terms of selectivity and yield. Using this auxiliary under the optimized reaction conditions allows for nearly complete stereoselectivity in a number of cyclic enol ether systems (93–95). Due to the extraordinary selectivity observed with this auxiliary, further investigations regarding the effect of zinc iodide on rate and selectivity were not preformed.
Once again, careful conformational analysis of the auxiliary provides an explanation for the observed stereochemical outcome (Scheme 3.27) [58]. First, the diol auxiliary can adopt a chair-like, six-membered ring chelate with the zinc carbenoid. Because of the anti orientation of the two isopropyl groups in the diol backbone, one is forced to reside in an axial position on the chelate ring. Assuming that the cyclohexene ring takes up an equatorial position, two limiting reactive conformations can then be drawn. In one case, xi, the cyclohexene ring is parallel to the plane of the zinc chelate ring. Approach of the methylene unit to the bottom (re) face of the alkene yields the observed, major diastereomer. In the other case the cyclohexene ring adopts a perpendicular orientation relative to the chelate ring as shown in xii. Here, the carbenoid must approach the top (si) face of the alkene. Cyclopropanation via this pathway would then lead to the minor diastereomer. Thus the stereochemical outcome is controlled by the preferred orientation of the cyclohexene ring (xi) which in turn can be understood in terms of the unfavorable syn pentane type interactions between the forming sp3 centers and the axial isopropyl group of the auxiliary as in xii.
In this model, the intermediacy of a monomeric zinc species is postulated. To support this assumption, an examination of the effect of stoichiometry and solvent in cyclopropanation involving the 2,4-pentanediol auxiliary was preformed [59]. In the initial reaction protocol, a large excess of both diethylzinc and diiodomethane is employed. Such excessive conditions are justified on account of the instability of the zinc carbenoid under the reaction conditions. To minimize the un-

114 3 Enantioselective [2+1] Cycloaddition: Cyclopropanation with Zinc Carbenoids
Scheme 3.27
productive process and reveal the most efficient stoichiometry of reagents, the diiodomethane is added to the mixture slowly. This reduces the amount of free carbenoid in solution and thus reduces any decomposition that may be occurring. This empirical survey of reagent stoichiometries demonstrates that a 4 : 3 ratio of diethylzinc to diiodomethane is required to obtain optimal reactivity and selectivity. The authors propose that under these conditions, in which the zinc carbenoid is still in a large excess as compared to the substrate alkene, are required due to reagent deactivation through strong solvation with the ethereal solvent.
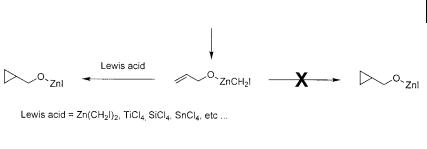
These conditions are quite unusual and deserve further comment. What is particularly interesting is that the diethylzinc is used in molar excess when compared to diiodomethane. The first equivalent of diethylzinc is required to deprotonate the free alcohol, thus forming the ethylzinc alkoxide (ROZnEt) 96 which can then react with diiodomethane to yield the iodomethylzinc alkoxide 97 (Scheme 3.28). Alone, it is unlikely that this species is active in the cyclopropanation. In a careful 1H NMR study, Charette et al. formed an iodomethylzinc alkoxide and observed that in the absence of added organozinc reagent, it was incapable of cyclopropanating the neighboring alkene (Scheme 3.29) [60]. However, the addition of any number of Lewis acidic metal species, including organozinc compounds, could revive the reactivity of the iodomethylzinc alkoxide.
Scheme 3.28
In light of this fact, one can now propose a role for the excess in the organozinc agents used by Tai. Three organozinc species remain after formation of the iodomethylzinc alkoxide 97 – diethylzinc, EtZnCH2I and Zn(CH2I)2. Coordination of any of these Lewis acidic species to the iodomethylzinc alkoxide should allow for the cyclopropanation to proceed through a species such as 98. This novel bimetallic species forms the putative reactive assembly. The rationale for the added reagents is revealed by later experiments. When a 1:1:1 mixture of diethylzinc, diiodomethane,
3.4 Stereoselective Simmons-Smith Cyclopropanations 115
Scheme 3.29
and the chiral enol ether is combined, no reaction is observed. Treatment of this solution with another equivalent of diethylzinc then leads to the formation of the product in a 98 : 2 dr and a 42% yield. This result agrees with the mechanistic proposal of a bimetallic, active cyclopropanating species similar to 98. However, this analysis only accounts for two equivalents of diethylzinc and possibly two equivalents of diiodomethane. Further explanation of the use of reagents beyond this ratio can only be accounted for in light of the aforementioned reagent decomposition.
Although the rationalization of the reactivity and selectivity of this particular substrate is distinct from that for chiral ketals 92–95, it still agrees with the mechanistic conclusions gained throughout the study of Simmons-Smith cyclopropanations. Still, the possibility of the existence of a bimetallic transition structure similar to v (see Fig. 3.5) has not been rigorously ruled out. No real changes in the stereochemical rationale of the reaction are required upon substitution of such a bimetallic transition structure. But as will be seen later, the effect of zinc iodide on catalytic cyclopropanations is a clue to the nature of highly selective reaction pathways. A similar but unexplained effect of zinc iodide on these cyclopropanation may provide further information on the true reactive species.
In addition to providing access to enantiomerically enriched cyclopropane-con- taining compounds, the auxiliary-direct reactions provide important insights into Simmons-Smith type cyclopropanations [61]. The success of conformational analysis in predicting the reactivity patterns for these substrates is a major advance. One similarity between these disparate methods is that they all rely upon the idea of a bimetallic transition structure. The role of the bridging zinc species (as in v) will take on greater significance in subsequent discussions of chirally modified reagents and catalytic methods.
3.4.3
In-situ Chiral Modification
3.4.3.1 Chirally Modified Reagents
An alternative approach to asymmetric synthesis that avoids covalent modification of the substrate is chiral modification of the active reagent. This not only streamlines the number of synthetic manipulations, but it simplifies the isolation of the desired product. In the case of zinc carbenoids, such modifications are feasible alternatives to the use of a standard chiral auxiliary. Two important factors combine

116 3 Enantioselective [2+1] Cycloaddition: Cyclopropanation with Zinc Carbenoids
to render this approach viable. First, the success of chirally modified zinc reagents in other reactions should be applicable to cyclopropanation with zinc carbenoids. Most notable among these reactions is the use of complexes of chiral amino alcohols and diethylzinc for the asymmetric alkylation of aldehydes [62]. Second, consideration of Rickborn’s proposed transition state assembly v in Fig. 3.5 reveals that the ZnI2 fragment in the bimetallic structure is acting as a spectator. It serves no other role than to bridge the oxygen atom and the active carbenoid. A chirally modified zinc reagent could be reasonably substituted in this position and provide a dissymmetric environment in close proximity to the alkene. On the basis of these factors it seems likely that chiral reagent modification could be successful in the Simmons-Smith cyclopropanation.
In recent years, several accounts of such chiral reagent modifications in cyclopropanation have appeared beginning with an early report by Fujisawa et al. who first demonstrated the concept and its potential (Table 3.6) [63]. Addition of Zn(CH2I)2 to a preformed zinc alkoxide 100 yields an enantiomerically enriched cyclopropane product 24. The experimental procedure illustrates how the chirally modified reagent participates in the cyclopropanation. First, the cinnamyl alcohol, diethyl tartrate and diethylzinc are combined in ratio that will provide the bis alkoxide. This bis-heteroatom-substituted zinc species can then interact with the active zinc carbenoid much in the same way as ZnI2. Now, however, the bridging zinc carries a chiral ligand, introducing a chiral environment in the methylene transfer step. Overall, yield and selectivity are moderate. The selectivity of the process is clearly sensitive to the steric bulk of the ester group. Whereas the ester ligands containing linear alkyl groups such as methyl (99a), ethyl (99b) and n-butyl (99c) react with comparable selectivities (approx. 75:25 dr), the bulky isopropyl ester (99d) shows a dramatic decrease in selectivity. It is interesting to note that these reactions proceed extremely slowly, requiring almost two days at –12 C to reach complete conversion. Similarly, slow reactions (ca. 24 h) were observed in the BINOL-modified, Simmons-Smith type cyclopropanations explored by Katsuki
Tab. 3.6 Tartrate reagent modification in the Simmons-Smith cyclopropanation
Entry |
Ester |
R |
Solvent |
Temp. ( C) |
Yield (%) |
er |
(24) |
|
|
|
|
|
|
|
|
1 |
99a |
Me |
CH2Cl2 |
0 to RT |
12 |
82 |
: 18 |
2 |
99b |
Et |
CH2Cl2 |
0 to RT |
22 |
75 |
: 25 |
3 |
99b |
Et |
(CH2Cl)2 |
–12 |
54 |
90 |
: 10 |
4 |
99c |
n-Bu |
CH2Cl2 |
0 to RT |
17 |
79 |
: 21 |
5 |
99d |
i-Pr |
CH2Cl2 |
0 to RT |
24 |
64 |
: 36 |
|
|
|
|
|
|
|
|

3.4 Stereoselective Simmons-Smith Cyclopropanations 117
Tab. 3.7 N-Methylephedrine reagent modification in Simmons-Smith cyclopropanation
Entry |
Solvent |
GC Conv. (%) |
Yield (%) |
er (24) |
|
|
|
|
|
|
|
1 |
hexane |
< 10 |
nd |
nd |
|
2 |
toluene |
> 90 |
82 |
59 : 41 |
(R,R) |
3 |
THF |
> 90 |
81 |
55 : 45 |
(S,S) |
4 |
DME |
> 90 |
85 |
62 : 38 |
(R,R) |
|
|
|
|
|
|
et al. [34 f ]. However, these reactions are able to provide high selectivities when compared to Fujisawa’s method.
Another method which is in line with the strategy of modifying the spectator zinc species was explored by Denmark et al. (Table 3.7) [64]. The amino alcohol N-methyl- ephedrine 101 is a potent ligand for asymmetric aldehyde additions. The zinc-ligand complex 102 is preformed before addition of the alcohol. The Zn(CH2I)2 is the final reagent added to the mixture and the addition order is similar to that used by Fujisawa. This reagent combination is able to provide the products in moderate yields and selectivities. Although high selectivity is never achieved, even under a variety of conditions, this result is still important because it supports the concept of a bimetallic transition structure with an inactive bridging zinc species.
All chiral reagent modifications do not rely upon an inactive, chiral zinc species. In 1998, Shi et al. undertook an investigation of a structurally unique, chirally modified reagent [23]. Rather than form a chiral zinc species and use it in the place of the bridging ZnI2, the active cyclopropanating agent is modified directly. By mixing one equivalent of an alcohol with Zn(CH2I)2, an iodomethylzinc alkoxide is presumed to form. Following the Charette analogy, the iodomethylzinc alkoxide must be activated by a Lewis acid, which in this case is diethylaluminum chloride. In the case of the fructose-derived alcohol 103 a 75:25 er is obtained for 106, notably with trans -methylstyrene, a simple alkene (Scheme 3.30).
As part of this study, Shi et al. noted that the reactivity of this oxygen-substi- tuted zinc carbenoid is considerably diminished with respect to the parent zinc species (Fig. 3.8). In the absence of an alcohol ligand (A), this protocol leads to a sluggish reaction (approx. 40% conversion after 10 h). This is not surprising in the case of an unfunctionalized alkene [7b]. The addition of some alcohol ligands, such as ethanol (B) (pKa = 15.9), 2-chloroethanol (B) (pKa = 14.3) and 2,2-dichloro- ethanol C (pKa = 12.9), lead to a suppression of the intrinsic reactivity of the carbenoid [65]. Only weakly basic oxygen containing groups, such as 2,2,2-trifluoro- ethanol D (pKa = 12.4), trifluoroacetate E (pKa 0.5) or benzoate F (pKa = 4.20), re-