

Cycloaddition Reactions in Organic Synthesis
.pdf178 4 Catalytic Enantioselective Cycloaddition Reactions of Carbonyl Compounds
4.3.4
Inverse Electron-demand Reactions
A simple approach for the formation of 2-substituted 3,4-dihydro-2H-pyrans, which are useful precursors for natural products such as optically active carbohydrates, is the catalytic enantioselective cycloaddition reaction of , -unsaturated carbonyl compounds with electron-rich alkenes. This is an inverse electron-demand cycloaddition reaction which is controlled by a dominant interaction between the LUMO of the 1- oxa-1,3-butadiene and the HOMO of the alkene (Scheme 4.2, right). This is usually a concerted non-synchronous reaction with retention of the configuration of the dienophile and results in normally high regioselectivity, which in the presence of Lewis acids is improved and, furthermore, also increases the reaction rate.
The inverse electron-demand catalytic enantioselective cycloaddition reaction has not been investigated to any great extent. Tietze et al. published the first example of this class of reaction in 1992 – an intramolecular cycloaddition of heterodiene 42 catalyzed by a diacetone glucose derived-titanium(IV) Lewis acid 44 to give the cis product 43 in good yield and up to 88% ee (Scheme 4.31) [46].
Scheme 4.31
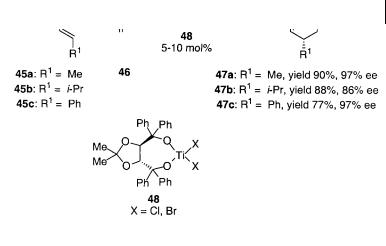
A chiral titanium(IV) complex has also been used by Wada et al. for the intermolecular cycloaddition of (E)-2-oxo-1-phenylsulfonyl-3-alkenes 45 with enol ethers 46 using the TADDOL-TiX2 (X = Cl, Br) complexes 48 as catalysts in an enantioselective reaction giving the dihydropyrans 47 as shown in Scheme 4.32 [47]. The reaction depends on the anion of the catalyst and the best yield and enantioselectivity were found for the TADDOL-TiBr2; up to 97% ee of the dihydropyrans 47 was obtained.
4.3 Cycloaddition Reactions of Carbonyl Compounds 179
Scheme 4.32
The chiral BOX-copper(II) complexes are effective catalysts for enantioselective cycloaddition reactions of , -unsaturated acyl phosphonates [48] and , -unsatu- rated keto esters [38b, 49].
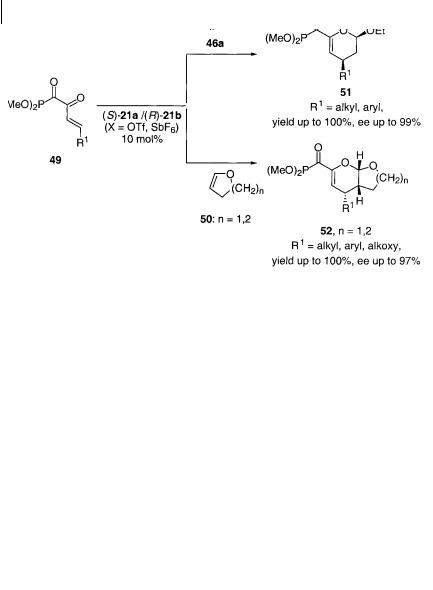
The chiral BOX-copper(II) complexes, (S)-21a and (R)-21b (X = OTf, SbF6), were found by Evans et al. to catalyze the enantioselective cycloaddition reactions of the, -unsaturated acyl phosphonates 49 with ethyl vinyl ether 46a and the cyclic enol ethers 50 giving the cycloaddition products 51 and 52, respectively, in very high yields and ee as outlined in Scheme 4.33 [38b]. It is notable that the acyclic and cyclic enol ethers react highly stereoselectively and that the same enantiomer is formed using (S)-21a and (R)-21b as the catalyst. It is, furthermore, of practical importance that the cycloaddition reaction can proceed in the presence of only 0.2 mol% (R)-21a (X = SbF6) with minimal reduction in the yield of the cycloaddition product and no loss of enantioselectivity (93% ee).
More recently, further developments have shown that the reaction outlined in Scheme 4.33 can also proceed for other alkenes, such as silyl-enol ethers of acetophenone [48 b], which gives the endo diastereomer in up to 99% ee. It was also shown that -ethyl- -methyl-substituted acyl phosphonate also can undergo a diastereoand enantioselective cycloaddition reaction with ethyl vinyl ether catalyzed by the chiral Ph-BOX-copper(II) catalyst. The preparative use of the cycloaddition reaction was demonstrated by performing reactions on the gram scale and showing that no special measures are required for the reaction and that the dihydropyrans can be obtained in high yield and with very high diastereoand enantioselective excess.
Our development of the catalytic enantioselective inverse electron-demand cycloaddition reaction [49], which was followed by related papers by Evans et al. [38, 48], focused in the initial phase on the reaction of mainly , -unsaturated -keto esters 53 with ethyl vinyl ether 46a and 2,3-dihydrofuran 50a (Scheme 4.34).
The enantioselective cycloaddition of the , -unsaturated -keto esters 53 with ethyl vinyl ether 46a and 2,3-dihydrofuran 50a catalyzed by 21b (X = OTf) or the
180 4 Catalytic Enantioselective Cycloaddition Reactions of Carbonyl Compounds
Scheme 4.33
Scheme 4.34
water complex 27 (Scheme 4.24) proceeds with high yield and diastereoand enantioselectivity [38, 48, 49]. The reaction tolerates a broad range of R1 substituents, such as alkyl, aryl, alkoxy, and thiobenzyl. The reaction can proceed with only 0.5 mol% catalyst 27b with only a slight decrease in ee and diastereomeric excess [38b, 48b]. Preliminary studies have also indicated that catalyst 27b can be re-used in multiple reaction cycles without significant loss in yield and stereoselectivity, and that the , -unsaturated -keto esters were somewhat more reactive than , - unsaturated acyl phosphonates in catalytic cycloaddition reactions.
4.3 Cycloaddition Reactions of Carbonyl Compounds 181
The absolute configuration of the cycloaddition products 54a,b was assigned by transformation to the lactones 66a,b with known configuration (Scheme 4.35) [38 b, 48 b].
Scheme 4.35
Further developments of this inverse electron-demand catalytic enantioselective cycloaddition reaction using -substituted , -unsaturated -keto esters 53 have resulted in simple approaches for the synthesis of optically active carbohydrates [39], including amino sugars [50], as outlined in Scheme 4.36. The reaction can be used for the preparation of optically active spiro carbohydrates 57, an important functionality found in natural compounds such a pheromones, steroidal compounds, antiparasitic agents, and polyether antibiotics. cis-Alkenes are also useful substrates for inverse electron-demand catalytic enantioselective cycloaddition reaction of , -unsaturated -keto esters leading to the cycloaddition product in good yield and with very high de and ee. This reaction has been used for the synthesis of the ethyl -d-manonose tetraacetate 58. It is interesting to point out that the - linkage at C-1 of the monosaccharides is difficult to synthesize by standard carbohydrate chemistry, because it is neither possible to use the neighboring-group effect at C-2, nor the anomeric effect. The cycloaddition approach can also be used as a synthetic procedure for the preparation of the non-naturally occurring acetalprotected C-2-branched carbohydrate 59. The formation of amino sugars by this catalytic enantioselective reaction has shown that diastereomers 60 and 61, with different protection groups, can be formed in high yield, de, and ee. Amino sugars can be applied as pharmaceuticals such as for the treatment of diabetes and are promising drugs against influenza.
The absolute configuration of products obtained in the highly stereoselective cycloaddition reactions with inverse electron-demand catalyzed by the t-Bu-BOX- Cu(II) complex can also be accounted for by a square-planar geometry at the copper(II) center. A square-planar intermediate is supported by the X-ray structure of the hydrolyzed enone bound to the chiral BOX-copper(II) catalyst, shown as 29b in Scheme 4.24.

182 4 Catalytic Enantioselective Cycloaddition Reactions of Carbonyl Compounds
Scheme 4.36
4.4
Summary
The major developments of catalytic enantioselective cycloaddition reactions of carbonyl compounds with conjugated dienes have been presented. A variety of chiral catalysts is available for the different types of carbonyl compound. For unactivated aldehydes chiral catalysts such as BINOL-aluminum(III), BINOL-tita- nium(IV), acyloxylborane(III), and tridentate Schiff base chromium(III) complexes can catalyze highly diastereoand enantioselective cycloaddition reactions. The mechanism of these reactions can be a stepwise pathway via a Mukaiyama aldol intermediate or a concerted mechanism. For -dicarbonyl compounds, which can coordinate to the chiral catalyst in a bidentate fashion, the chiral BOX-copper(II)

References 183
complexes have shown promising results. These complexes can catalyze cycloaddition reactions of glyoxylates, -keto esters, -diketones, and ketomalonate with conjugated dienes, leading to the cycloaddition product in high yield, diastereoand enantioselectivity. Occasionally it was possible to perform the reactions with low catalyst loading. These normal electron-demand reactions proceed via different structural intermediates depending on the chiral ligand. The chiral BOX-cop- per(II) complexes can also be used as catalysts for inverse electron-demand cycloaddition reactions of , -unsaturated carbonyl compounds with electron-rich alkenes leading to a simple procedure for the formation 2-substituted 3,4-dihydro- 2H-pyrans. These reactions also proceed in a highly selective manner and have been used for the synthesis of carbohydrates such as spiro carbohydrates, a -d- manonose, and amino sugars.
Acknowledgment
This work was made possible by a grant from The Danish National Research Foundation.
References
[1] |
O. Diels, K. Alder, Liebigs Ann. Chem. |
|
ing, Frontier Orbitals and Organic Chemi- |
|
1928, 460, 98. |
|
cal Reactions, John Wiley & Sons: London |
[2] |
See e.g.: (a) W. Carruthers, Cycloaddi- |
|
1977. |
|
tion Reactions in Organic Synthesis, Tetra- |
[5] |
See e.g.: J. Juracak, A. Golebiowski, A. |
|
hedron Organic Chemistry Series Vol. 8; |
|
Rahm, Tetrahedron Lett. 1986, 27, 853. |
|
Pergamon Press: Elmsford, NY 1990; (b) |
[6] |
S. J. Danishefsky, H. G. Selnick, R. E. |
|
I. Ojima, Catalytic Asymmetric Synthesis, |
|
Zelle, M. P. DeNinno, J. Am. Chem. |
|
VCH Publishers, Inc.: New York, 1993; |
|
Soc. 1988, 110, 4368. |
|
(c) R. Noyori, Asymmetric Catalysis in Or- |
[7] |
S. J. Danishefsky, E. Larson, D. Ash- |
|
ganic Synthesis, John Wiley: New York, |
|
kin, N. Kato, J. Am. Chem. Soc. 1985, |
|
1994; (d) K. C. Nicolaou, E. J. Sorensen, |
|
107, 1246. |
|
Classics in Total Synthesis Targets, Strategies, |
[8] |
See e.g.: (a) K. Mikami, M. Terada, T. |
|
Methods, VCH publishers, Inc.: New York, |
|
Nakai, J. Am. Chem. Soc. 1990, 112, |
|
1996; (e) D. L. Boger, S. H. Weinreb, |
|
3949; (b) K. Mikami, M. Shimizu, Chem. |
|
Hetero-Diels-Alder Methodology in Organic |
|
Rev. 1992, 92, 1021; (c) D. J. Berrisford, |
|
Synthesis, Academic Press: New York 1987; |
|
C. Bolm, Angew. Chem., Int. Ed. Engl. |
|
(f) H. Waldmann, Synthesis 1994, 535; (g) |
|
1995, 34, 1717. |
|
L. F. Tietze, G. Kettschau, Top. Curr. |
[9] |
M. Johannsen, K. A. Jørgensen, J. Org. |
|
Chem. 1997, 190, 1; (h) L. F. Tietze, Curr. |
|
Chem. 1995, 60, 5757. |
|
Org. Chem. 1998, 2, 19. |
[10] |
A. Graven, M. Johannsen, K. A. |
[3] |
Recent reviews dealing with catalytic en- |
|
Jørgensen, J. Chem. Soc., Chem. Com- |
|
antioselective cycloaddition reactions of |
|
mun. 1996, 2373. |
|
carbonyl compounds and imines see, |
[11] |
(a) K. Maruoka, T. Itoh, Y. Araki, T. |
|
e.g., Refs. 2(g) and 2(h); K. A. |
|
Shirasaka, H. Yamamoto, Bull. Chem. |
|
Jørgensen, Angew. Chem. Int. Ed. 2000, |
|
Soc. Jpn. 1988, 61, 2975; (b) K. Maruoka, |
|
39, 3558. |
|
Y. Hoshino, T. Shirasaka, H. Yamamo- |
[4] |
See e.g.: R. B. Woodward, R. Hoff- |
|
to, Tetrahedron Lett. 1988, 29, 3967; (c) K. |
|
mann, The Conservation of Orbital Sym- |
|
Maruoka, T. Itoh, T. Shirasaka, H. Ya- |
|
metry; Verlag Chemie 1970; (b) L. Flem- |
|
mamoto, J. Am. Chem. Soc. 1988, 110, |

184 4 Catalytic Enantioselective Cycloaddition Reactions of Carbonyl Compounds
310; (d) K. Maruoka, A. B. Concepcion,
H.Yamamoto, Bull. Chem. Soc. Jpn.
1992, 65, 3501.
[12]K. Maruoka, H. Yamamoto, J. Am. Chem. Soc. 1989, 111, 789.
[13]K. B. Simonsen, N. Svenstrup, M. Roberson, K. A. Jørgensen, Chem. Eur. J.
2000, 6, 123.
[14]M. Roberson, A. S. Jepsen, K. A. Jørgensen, Tetrahedron 2001, 57, 907.
[15]L.-Z. Gong, L. Pu, Tetrahedron Lett. 2000,
41, 2327.
[16]M. Quimpère, K. Jankowski, J. Chem. Soc., Chem. Commun. 1987, 676.
[17](a) Q. Gao, T. Maruyama, M. Mouri, H. Yamamoto, J. Org. Chem. 1992, 57, 1951;
(b) Q. Gao, K. Ishihars, T. Maruyama,
M.Mouri, H. Yamamoto, Tetrahedron
1994, 50, 979.
[18](a) M. Takasu, H. Yamamoto, Synlett 1990, 194; (b) D. Sartor, J. Saffrich, G. Helmchen, Synlett 1990, 197; (c) D. Sartor, J. Saffrich, G. Helmchen, C. J. Richards, H. Lambert, Tetrahedron: Asymmetry 1991, 2, 639; (d) E. J. Corey, T.-P. Loh, J. Am. Chem. Soc. 1991, 113, 8966;
(e) E. J. Corey, T.-P. Loh, T. D. Roper, M.
D.Azimioara, M. C. Noe, J. Am. Chem. Soc. 1992, 114, 8290; (f) E. J. Corey, A. Guzman-Perez, T.-P. Loh, J. Am. Chem. Soc. 1994, 116, 3611; (g) E. J. Corey, T.-P. Loh, Tetrahedron Lett. 1993, 34, 3979; (h)
J.Corey, C. L. Cywin, T. D. Roper, Tetrahedron Lett. 1992, 33, 6907.
[19]See also: (a) K. Mikami, S. Matsukawa,
J.Am. Chem. Soc. 1993, 115, 7039; (b) K.
Mikami, Y. Motoyama, M. Terada, J. Am. Chem. Soc. 1994, 116, 2812; (c) K. Mikami, S. Matsukawa, Nature 1997, 385, 613; (d) G. E. Keck, K. H. Tarbet, L. S. Geraci, J. Am. Chem. Soc. 1993,
115, 8467; (e) G. E. Keck, D. Krishnamurthy, J. Am. Chem. Soc. 1995, 117, 2363; (f) G. E. Keck, X.-Y. Li, D. Krishnamurthy, J. Org. Chem. 1995, 60, 5998;
(g) K. Maruoka, N. Murase, H. Yamamoto, J. Org. Chem. 1993, 58, 2938; (h)
A. L. Costa, M. G. Piazza, E. Tagliavini, C. Trombini, A. Umani-Ronchi, J. Am. Chem. Soc. 1993, 115, 7001; (i) R. O.
Duthaler, A. Hafner, Angew. Chem., Int. Ed. Engl. 1997, 36, 43.
[20]L. Lévëque, M. Le Blanc, R. Pastor, Tetrahedron Lett. 2000, 41, 5043.
[21]A. Togni, Organometallics 1990, 9, 3106.
[22]S. E. Schaus, J. Brånalt, E. N. Jacobsen,
J.Org. Chem. 1998, 63, 403.
[23]A. G. Dossetter, T. F. Jamison, E. N. Jacobsen, Angew. Chem. Int. Ed. 1999, 38, 2398.
[24](a) M. Bednarski, C. Maring, S. Danishefsky, Tetrahedron Lett. 1983, 24, 3451; (b) M. Bednarski, S. Danishefsky, J. Am. Chem. Soc. 1983, 105, 6968; (c)
J.Am. Chem. Soc. 1986, 108, 7060.
[25]T. Hanamoto, H. Furuno, Y. Sugimoto,
J.Inanaga, Synlett 1997, 79.
[26]M. Johannsen, K. A. Jørgensen, Z.-M. Lin, Q.-S. Hu, L. Pu, J. Org. Chem. 1999,
64, 299.
[27]Y. Motoyama, K. Mikami, J. Chem. Soc., Chem. Commun. 1994, 1563.
[28](a) K. Mikami, M. Terada, Y. Motoyama,
T.Nakai, Tetrahedron: Asymmetry 1991, 2, 643; (b) S. Matsukawa, K. Mikami, Tetrahedron: Asymmetry 1997, 8, 815; (c) Y. Motoyama, M. Terada, K. Mikami, Synlett 1995, 967.
[29](a) Y.-J. Hu, X.-D. Huang, Z.-J. Yao, Y.-L. Wu, J. Org. Chem. 1998, 63, 2456; (b) L.-
S.Li, Y. Wu, Y.-J. Hu, L.-J. Xia, Y.-L. Wu,
Tetrahedron: Asymmetry 1998, 9, 2271.
[30]See e.g.: (a) A. Pfaltz, Acc. Chem. Res.
1993, 26, 339; (b) A. K. Ghosh, P. Mathivanan, J. Cappiello, Tetrahedron: Asymmetry 1998, 9, 1; (c) K. A. Jørgensen, M. Johannsen, S. Yao, H. Audrain, J. Thorhauge, Acc. Chem. Res. 1999, 32, 605; J. S. Johnson, D. A. Evans, Acc. Chem. Res. 2000, 33, 325.
[31]For the application of C2-symmetric bis- oxazoline-Lewis acids in other catalytic reactions: (a) Mukaiyama-aldol reactions see, e.g., D. A. Evans, M. C. Kozlowski,
J.A. Murry, C. S. Burgey, K. R. Campos,
B.T. Connell, R. J. Staples, J. Am. Chem. Soc. 1999, 121, 669 and references therein; D. A. Evans, C. S. Burgey, M. C. Kozlowski, S. W. Tregay, J. Am. Chem. Soc. 1999, 121, 686 and references therein; (b) Diels-Alder reactions see e.g.:
D.A. Evans, D. M. Barnes, J. S. Johnson, T. Lectka, P. v. Matt, S. J. Miller,
R.D. Norcross, E. A. S. haughnessy,
K.R. Campos, J. Am. Chem. Soc. 1999,
121, 7582 and references cited therein;

D. A. Evans, S. J. Miller, T. Lectka, P. von Matt, J. Am. Chem. Soc. 1999, 121, 7559 and references therein; (c) 1,3-Dipo- lar cycloaddition reactions see e.g.: K. V. Gothelf, R. G. Hazell, K. A. Jørgensen,
J. Org. Chem. 1996, 61, 346; K. B. Jensen,
R.G. Hazell, K. A. Jørgensen, J. Org. Chem. 1999, 64, 2353; (d) cyclopropanation reactions see e.g.: R. E. Lowenthal,
S.Masamune, Tetrahedron Lett. 1991, 32, 7373; D. A. Evans, K. A. Worpel, M. M. Hinman, M. M. Faul, J. Am. Chem. Soc. 1991, 113, 726; A. Evans, K. A. Woerpel,
M.J. Scott, Angew. Chem. Int. Ed. Engl.
1992, 31, 430; T. G. Gant, M. C. Noe, E. J.
Corey, Tetrahedron Lett. 1995, 36, 8745;
(e) allylic substitution reactions see e.g.:
P. von Matt, G. C. Lloyd-Jones, A. B. E. Minidis, A. Pfaltz, L. Macko, M. Neuburger, M. Zehnder, H. Rüegger, P. S. Pregogin, Helv. Chim. Acta 1995, 78, 265; (f) allylation and addition reactions see e.g.: J. H. Wu, R. Radinov, N. A. Porter, J. Am. Chem. Soc. 1995, 117, 11029;
M.P. Sibi, J. Ji, J.-H. Wu, S. Gurtler,
N.A. Porter, J. Am. Chem. Soc. 1996,
118, 9200; D. A. Evans, T. Rovis, M. C. Kozlowski, J. S. Tedrow, J. Am. Chem. Soc. 1999, 121, 1994; (g) aziridination reactions see e.g.: D. A. Evans, M. M. Faul,
M.T. Bilodeau, B. A. Anderson, D. M. Barnes, J. Am. Chem. Soc. 1993, 115, 5328; K. B. Hansen, N. S. Finney, E. N. Jacobsen, Angew. Chem., Int. Ed. Engl.
1995, 34, 676; (h) carbonyl-ene reactions see e.g.: D. A. Evans, C. S. Burgey, N. A.
Paras, T. Vojkovsky, S. W. Tregay, J. Am. Chem. Soc. 1998, 120, 5824; F. Reichel,
X.Fang, S. Yao, M. Ricci, K. A. Jørgensen, Chem. Commun. 1999, 1505;
N.Gathergood, K. A. Jørgensen,
Chem. Commun. 1999, 1869.
[32]M. Johannsen, K. A. Jørgensen, Tetrahedron 1996, 52, 7321; (b) J. Chem. Soc., Perkin Trans. 2 1997, 1183; (c) M. Johannsen, S. Yao, A. Graven, K. A. Jørgensen, Pure Appl. Chem. 1997, 70, 1117.
[33]S. Yao, M. Johannsen, R. G. Hazell,
K.A. Jørgensen, J. Org. Chem. 1998, 63, 118.
[34]A. K. Ghosh, P Mathivanan, J. Cappiello, K. Krishnan, Tetrahedron: Asymmetry
1996, 7, 2165.
References 185
[35]A. K. Ghosh, P Mathivanan, J. Cappiello, Tetrahedron Lett. 1997, 38, 2427.
[36]S. Yao, M. Johannsen, K. A. Jørgensen,
J.Chem. Soc., Perkin Trans. 1 1997, 2345.
[37]G. Desimoni, G. Faita, P. P. Righetti,
N.Sardone, Tetrahedron 1996, 52, 12019.
[38](a) D. A. Evans, J. S. Johnson, C. S.; Burgey, C. R. Campos, K. R. Tetrahedron Lett. 1999, 40, 2879; (b) D. A. Evans, E. J. Olhava, J. S. Johnson, J. M. Janey, Angew. Chem. Int. Ed. 1998, 37, 3373.
[39]H. Audrian, J. Thorhauge, R. G. Hazell, K. A. Jørgensen, J. Org. Chem,
2000, 65, 4487.
[40]D. A. Evans, S. J. Miller, T. Lectka, J. Am. Chem. Soc. 1993, 115, 6460.
[41](a) S. Oi, K. Kashiwagi, E. Terada, K. Ohuchi, Y. Inoue, Tetrahedron Lett.
1996, 37, 6351; (b) S. Oi, E. Terada, K. Ohuchi, T. Kato, Y. Tachibana, Y. Inoue, J. Org. Chem, 1999, 64, 8660.
[42]K. Mikami, O. Kotera, Y. Motoyama, H. Sakaguchi, Synlett 1995, 975.
[43](a) M. Johannsen, S. Yao, K. A. Jørgensen, Chem. Commun. 1997, 2169;
(b) S. Yao, M. Johannsen, H. Audrain,
R.G. Hazell, K. A. Jørgensen, J. Am. Chem. Soc. 1998, 120, 8599.
[44]D. A. Evans, J. A. Murry, P. von Matt,
R.D. Norcross, S. J. Miller, Angew. Chem., Int. Ed. Engl. 1995, 34, 798.
[45]S. Yao, M. Roberson, F. Reichel, K. A. Jørgensen, J. Org. Chem. 1999, 64, 6677.
[46]L. F. Tietze, P. Saling, Synlett, 1992, 281; Chirality 1993, 5, 329.
[47](a) E. Wada, H. Yasuoka, S. Kanamasa,
Chem. Lett, 1994, 1637; (b) E. Wada, W. Pei, H. Yasuoka, U. Chin, S. Kanamasa,
Tetrahedron 1996, 52, 1205.
[48](a) D. A. Evans, J. S. Johnson, J. Am. Chem. Soc. 1998, 120, 4895; (b) D. A. Evans, J. S. Johnson, E. J. Olhava, J. Am. Chem. Soc. 2000, 122, 1635.
[49]J. Thorhauge, M. Johannsen, K. A. Jørgensen, Angew. Chem Int. Ed. 1998,
37, 2404.
[50]W. Zhuang, J. Thorhauge, K. A. Jørgensen, Chem. Commun. 2000, 459.
Cycloaddition Reactions in Organic Synthesis. 187
Edited by S. Kobayashi and K. A. Jorgensen
Copyright © 2001 Wiley-VCH Verlag GmbH
ISBNs: 3-527-30159-3 (Hardcover); 3-527-60025-6 (Electronic)
5
Catalytic Enantioselective Aza Diels-Alder Reactions
Shu Kobayashi
5.1
Introduction
Asymmetric aza Diels-Alder reactions provide a useful route to optically active ni- trogen-containing heterocyclic compounds such as piperidines, tetrahydroquinolines, etc. [1]. Although successful examples of diastereoselective approaches using chiral auxiliaries have been reported [2], few examples of enantioselective reactions are known. This is in remarkable contrast to the recent progress achieved in asymmetric Diels-Alder reactions of carbon dienophiles or hetero-Diels-Alder reactions of carbonyl dienophiles with dienes. Although chiral Lewis acids have played leading roles in these reactions, it is assumed that most Lewis acids would be trapped by basic nitrogen atoms of the starting materials and/or products in aza Diels-Alder reactions, and that truly catalytic reactions would be difficult to perform. As a pioneering effort, Yamamoto et al. reported enantioselective aza DielsAlder reactions of aldimines with Danishefsky’s diene using chiral boron compounds (Scheme 5.1) [3]. Although high enantioselectivity was obtained, stoichiometric amounts of chiral sources were needed. This is also true in the above ex-
Scheme 5.1

188 5 Catalytic Enantioselective Aza Diels-Alder Reactions
ample in which the chiral boron Lewis acids might be trapped by basic moieties of starting materials or products.
In 1996, the first example of the catalytic enantioselective aza Diels-Alder reactions of azadienes using a chiral lanthanide catalyst was reported [4]. In this article, successful examples of such catalytic reactions are surveyed.
5.2
Aza Diels-Alder Reactions of Azadienes
To achieve catalytic enantioselective aza Diels-Alder reactions, choice of metal is very important. It has been shown that lanthanide triflates are excellent catalysts for achiral aza Diels-Alder reactions [5]. Although stoichiometric amounts of Lewis acids are often required, a small amount of the triflate effectively catalyzes the reactions. On the basis of these findings chiral lanthanides were used in catalytic asymmetric aza Diels-Alder reactions. The chiral lanthanide Lewis acids were first developed to realize highly enantioselective Diels-Alder reactions of 2-oxazolidin-1- one with dienes [6].
The reaction of N-benzylideneaniline with cyclopentadiene was performed under the influence of 20 mol% of a chiral ytterbium Lewis acid prepared from ytterbium triflate (Yb(OTf)3) [7], (R)-(+)-1,1 -binaphthol (BINOL), and trimethylpiperidine (TMP). The reaction proceeded smoothly at room temperature to afford the desired tetrahydroquinoline derivative in 53% yield, although no chiral induction was observed. At this stage, it was indicated that bidentate coordination between a substrate and a chiral Lewis acid would be necessary for reasonable chiral induction. Thus, N-benzylidene-2-hydroxyaniline (1a) was chosen as an imine, and the reaction with cyclopentadiene (2a) was examined. It was found that the reaction proceeded smoothly to afford the corresponding 8-hydroxyquinoline derivative (3a) in high yield. It is noted that some interesting biological activities were reported in 8-hydroxyquinoline derivatives [8]. Although the enantiomeric excess of the cis adduct in the first trial was only 6%, the selectivity increased when diazabicyclo- [5,4,0]-undec-7-ene (DBU) was used instead of TMP (Scheme 5.2, Table 5.1). It was also indicated that the phenolic hydrogen of 1a would interact with DBU, which should interact with the hydrogen of (R)-(+)-BINOL [9], to reduce the selectivity. Several additives which interact with the phenolic hydrogen of 1a were then examined. When 20 mol% of N-methylimidazole (NMI) was used, 91% ee of the cis adduct was obtained, but the chemical yield was low. When other additives were screened it was found that the desired tetrahydroquinoline derivative was obtained in 92% yield with high selectivity (cis/trans = >99/1, 71% ee), when 2,6-di-t- butyl-4-methylpyridine (DTBMP, 4a) was used [4].
Other substrates were tested; the results are summarized in Table 5.2. Vinyl ethers (2b–2d) also worked well to afford the corresponding tetrahydroquinoline derivatives (3a–3e) in good to high yields with good to excellent diastereoand enantioselectivity (entries 1–10). Use of 10 mol% of the chiral catalyst also gave the adducts in high yields and selectivity (entries 2 and 6). As for additives, 2,6-di-t-bu-