

Astruc D. - Modern arene chemistry (2002)(en)
.pdf
55415 Oxidative Conversion of Arenols into ortho-Quinols and ortho-Quinone Monoketals
was achieved by using PIFA as a non-nucleophilic oxidizing agent in the presence of oxida- tion-sensitive allylsilane or 1-trimethylsilyloxybuta-1,3-diene as soft carbon nucleophiles [113, 114]. The trifluoroacetic acid (TFA) released during the oxidation step generates an acidic medium that is certainly propitious to the requisite cleavage of the SiaC and SiaO bonds. The naphthalenone 68a had previously been synthesized by Adler oxidation of 2-allylnaph- thol in a pitiful yield of 0.53 % [115]. In the present case, the 2-methoxy group acts as a powerful regioselector aiding in the elaboration of the carbon–carbon bond; no C-4 adduct 69a is observed. An unfortunate loss of selectivity is observed with the 2-benzyloxynaphthol 64b, however, probably because of the greater steric demand of the phenyl group. There exist a few other approaches for the preparation of ortho-quinol derivatives besides those that can be grouped within the aforementioned three main categories. Our recent review on this topic constitutes a source of leading references for these subsidiary methods [6].
15.3
Synthetic Applications of ortho-Quinols and ortho-Quinone Monoketals
The predisposition of ortho-quinols and ortho-quinone monoketals toward aromatizing rearrangement and dimerization is at the origin of the di culties that surround their manipulations in organic synthesis (Section 15.1.2.1) [1, 2]. Aromatization through a methyl 1,2- shift was, for example, the cause of failure of the ortho-quinol-based route initially adopted by Wood and co-workers in their recent synthesis of epoxysorbicillinol (17; Figure 6) [116]. We have already noted that ortho-quinol acetates are less inclined to undergo dimerization than ortho-quinone dialkyl ketals, as well as free ortho-quinols, but that they are quite sensitive to rearomatizing acetate shifts [117–119]. We experienced this rearrangement chemistry ourselves when submitting certain ortho-quinol acetates such as 62 to silica gel chromatography (vide supra). Pericyclic [3,3]- and ‘‘pseudopericyclic’’ [3,5]sigmatropic acetate shifts furnished the phenols 63a and 63b, respectively (Figure 19) [6, 80].
It is nevertheless possible to bridle the reactivity of ortho-quinols and ortho-quinone monoketals for rapid elaboration of structural complexity and diversity in a pertinent manner for target-oriented synthesis. Some remarkable accomplishments have been highlighted in several review articles [3, 5, 6, 120]. Among the most significant ones, one can cite rearrangements of bicyclo[2.2.2]octenone cycloadducts into decalins, ring-contractions to polyquinanes, ring-expansions to tropolones, intramolecular cycloadditions to isotwistanes, cationic annulations to benzofurans, and nucleophilic additions to biaryls, enediyne units, and benzannulated heterocycles of various sizes.
15.3.1
Diels–Alder Cycloadditions
The ability of the cyclohexa-2,4-dienone unit of ortho-quinone monoketals and other orthoquinol derivatives to react as either a diene or a dienophile component in [4pþ2p] cycloadditions is arguably their principal virtue in organic synthesis, and paradoxically it is also the principal reason why it is often di cult to exploit them in synthesis; they often dimerize faster than they can combine with another p-system partner. Adler, Andersson, and coworkers have extensively studied the behavior of ortho-quinols in Diels–Alder cycloadditions,

15.3 Synthetic Applications of ortho-Quinols and ortho-Quinone Monoketals 555
Fig. 21
and observed that systems bearing substituents at their 5-position do not dimerize as readily [85–93, 95, 121]. A substituent at this locus can apparently interfere with the geminal C-6 groups of the other unit in their endo-selective ‘‘back-to-back’’ mutual approach, which is observed in all reported cases [1, 6]. Thus, for example, 6-hydroxy-5,6-dimethyl- (70a) and 6- hydroxy-3,5,6-trimethylcyclohexa-2,4-dienone (70b) do not dimerize, whereas 6-hydroxy-4,6- dimethyl- (72a) and 6-hydroxy-2,4,6-trimethylcyclohexa-2,4-dienone (72b) do (Figure 21) [85, 94, 95]. An explanation for the exclusive and regioselective participation of the 4,5-double bond as the 2p partner in these dimerizations can be found in a report by Houk [122], who calculated that both the HOMO and the LUMO of 1-substituted electron-deficient dienes possess slightly higher atomic coe cients at the sp2-carbon center remote from the carbonyl group, that is C-5 in the dimerizing cyclohexa-2,4-dienone units [79]. Other calculations made on 1-substituted and 1,4-disubstituted dienes did not allow any clear-cut interpretation and hence brought the debate round to the influence of secondary orbital interactions [122– 124]. The regioselectivity in Diels–Alder dimerizations of ortho-quinol derivatives still awaits theoretical clarification.
This tendency toward spontaneous dimerization was further elaborated for ortho-quinone monoketals by Liao, who concluded that electron-withdrawing and/or small groups at the 4-position of 6,6-dimethoxycyclohexa-2,4-dienones tend to facilitate self-dimerization, whereas electron-releasing and/or large groups at their 2- and/or 4-positions, as well as small electron-releasing groups at their 5-position, have the reverse e ect [125]. A bromide in the 4-position is particularly e cient at retarding self-dimerization [126]. Liao and co-workers also suggested that one methoxy group in ortho-quinone dimethyl ketals participates in sec-


558 15 Oxidative Conversion of Arenols into ortho-Quinols and ortho-Quinone Monoketals
Fig. 25
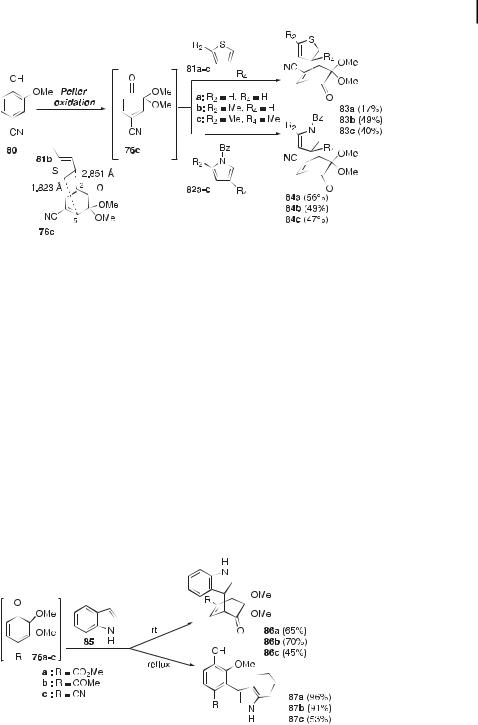
furan-, pyrrole-, thiophene-, and indole-derived bicyclo[2.2.2]octenones (Figures 22–24). These regiochemical observations are corroborated by those reported in Liao’s series of papers on the Diels–Alder reactivity between electron-rich dienophiles, such as benzyl vinyl ether 88b, dihydrofuran 89a, dihydropyran 89b, phenyl vinyl sulfide 88d, and styrene 88e, and ortho-quinone monoketals derived from eight di erent 2-methoxyphenols (Figure 25) [141, 142].
Calculations performed at the HF/3-21G level indicated smaller energy gaps between the HOMOs of the aforementioned electron-rich dienophiles and the LUMOs of the quinone ketals, as can be expected for inverse electron-demand Diels–Alder reactions under FMO control [141]. Regiochemical controls observed with quinone ketals such as 76a were well corroborated by the relative magnitudes of the atomic coe cients of the frontier orbitals. The highest coe cients at C-5 of the quinone ketal LUMO and at C-2 of the electron-rich alkenes would indeed promote bond formation between these centers. The results of calculations on other quinone ketals were, however, rather vague [141].
Numerous applications of the Diels–Alder reaction of ortho-quinone monoketals and ortho-quinols in natural products synthesis have been reported over the years [6]. The following few recent examples have been selected with the aim of illustrating the usefulness of the cycloaddition of ortho-quinol derivatives to give bicyclo[2.2.2]octenones, for these formidable strategic synthons can be further transformed in many ways to give various polycyclic architectures. Liao’s group has added a couple of notches to its already remarkable record in the field. They described a total synthesis of the norsesquiterpenoid (G)-eremopetasidione 97 [143], the key steps of which were the Diels–Alder reaction of the quinone ketal 93 with ethyl vinyl ketone, followed by a Cope rearrangement of the resulting bicyclo[2.2.2]octenone system 94 (Figure 26).
The first total synthesis of (G)-pallescensin 103 was also reported by Liao’s group [144] and features an intramolecular Diels–Alder reaction initiated by a regioselective oxidative alkoxylation of 2-methoxy-4-methylphenol 98 using the allylic alcohol 99 in the presence of PIDA. Vinylation of the resulting bicyclo[2.2.2]octenone 100, followed by ring-expansion through an ionic 1,3-allylic shift gave the bicyclo[4.2.2]decenone 102, which was further transformed into (G)-103 (Figure 27).



15.3 Synthetic Applications of ortho-Quinols and ortho-Quinone Monoketals 561
Fig. 30
15.3.2
Photochemical Rearrangements
Cyclohexa-2,4-dienones are particularly sensitive to photochemically-induced transformations. Seminal investigations by Barton and co-workers [158, 159] have demonstrated the facile thermally reversible photoisomerization of these linearly conjugated cyclohexadienones into dienyl ketenes, which can evolve into hexadienoic acid derivatives in the presence of a protonic nucleophile (NuH) (e.g. 126, Figure 31) [160, 161]. Reaction conditions favoring the p ,n excitation state as the lowest excited state are best for initiating such a ring-opening event. Under certain conditions allowing the p ,p excitation path to be followed, bicyclo[3.1.0]hexenones can be produced directly (e.g. 134, Figure 33), as first observed by Hart and co-workers [160–162]. Early on, ortho-quinol acetates were used as substrates to study the photochemical behavior of cyclohexa-2,4-dienones [158, 159], and numerous examples have been described by Quinkert and co-workers [160, 163, 164]. In contrast to the photolytic ring-opening of cyclohexa-2,4-dienones bearing two carbon substituents at their 6-position, ortho-quinol acetates of type 123 only led to diene ketenes with the (5E )-configuration (Figure 31).
This stereochemical preference is attributed to a stereoelectronic promotion of the C6–C7 bond into a pseudoaxial orientation due to a stabilizing interaction between the corresponding s orbital and the antibonding p CbO orbital in the photoexcited cyclohexadienone 124 (Figure 31). Quinkert additionally invoked a through-space interaction between the two polar carbonyl groups to rationalize the conformational preference leading to the (5E )-geometry [160]. From a general viewpoint, di erent primary and several secondary photoproducts can be obtained under di erent reaction conditions from di erently substituted starting orthoquinol acetates. Of particular note is the possibility of building seven-membered rings from ortho-quinol acetates such as 127a–c bearing a side chain with a 2-oxo group at the 6-position (Figure 32). The presence of a bulky substituent at the 4-position increases the deviation from planarity of the nucleophilically-trapped primary photoproduct 128a–c. This deviation
