

Astruc D. - Modern arene chemistry (2002)(en)
.pdf
584 16 Molecular Switches and Machines Using Arene Building Blocks
Fig. 13. The shuttling process and energy barrier for the passage of the tetracationic cyclophane between the two degenerate recognition sites, A and B, in the [2]rotaxane 214þ.
and another set arising from the protons on the free HQ ring. Indeed, at room temperature in [D6]acetone solution, the signals of the HQ protons are merged into the baseline between d ¼ 3:5 and d ¼ 6:5, suggesting that the shuttling process in the molecular shuttle 21 4PF6 is slow on the 1H NMR time-scale at room temperature. Cooling the solution of 21 4PF6 in [D6]acetone to 223 K in an e ort to further slow down the shuttling process allowed two sets of peaks to be observed at d ¼ 6:38 and d ¼ 3:8, corresponding to the protons on the free and encircled HQ rings, respectively. When the same sample was warmed up to 413 K in [D6]DMSO, only one set of proton signals was observed at d ¼ 5:16 because of the rapid shuttling process. By monitoring the various signal coalescences with di erent 1H NMR probes in both the dumbbell and macrocycle components, the energy barrier (DGz) to the shuttling process was determined [13a, 13e] to be in the range 12.4–13.3 kcal mol 1.
It is also possible to control the shuttling process within a non-degenerate molecular shuttle. By incorporating two recognition sites with di erent p-electron-donor/acceptor abilities into the dumbbell-shaped component, the macrocycle can be located preferentially on one of the two di erent recognition sites. The macrocycle can then be enticed to reside on the other recognition site by altering the p-electron-donor/acceptor ability of the original recognition site. The [2]rotaxane 224þ constitutes [14] a chemically and electrochemically switchable molecular shuttle. The synthesis of [2]rotaxane 224þ 4PF6 is illustrated in Figure 14. The bis(hexafluorophosphate) salt 6 2PF6 was treated with 1,4-bis(bromomethyl)benzene (7) in the presence of the dumbbell-shaped compound 23, which incorporates both benzidine and biphenol recognition sites. The chemically and electrochemically controllable switching relies upon the p-p stacking and [CaH O] interactions between the complementary units in the recognition sites. As expected, the [2]rotaxane 224þ exhibits translational isomerism, with the tetracationic cyclophane located preferentially (84:16 in MeCN at44 C) on the more p-electron-rich benzidine ring. Two translational isomers could also be detected by absorption spectroscopy. The absorption spectrum recorded in MeCN shows two

586 16 Molecular Switches and Machines Using Arene Building Blocks
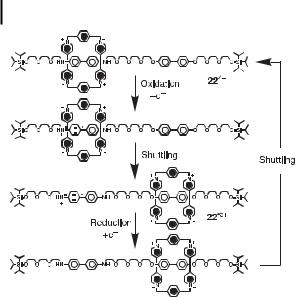
Fig. 16. The redox-controllable switching of the [2]rotaxane 22 4PF6.
Cyclic voltammetry shows that the [2]rotaxane 224þ can also undergo electrochemical switching (Figure 16) by a monoelectronic oxidation process to give the radical pentacationic species 22 5þ. On oxidation, the benzidine unit is converted to its monocationic radical state, generating an electrostatic repulsion, which causes the tetracationic cyclophane to move to the biphenol unit in [2]rotaxane 224þ. This redox procedure is completely reversible.
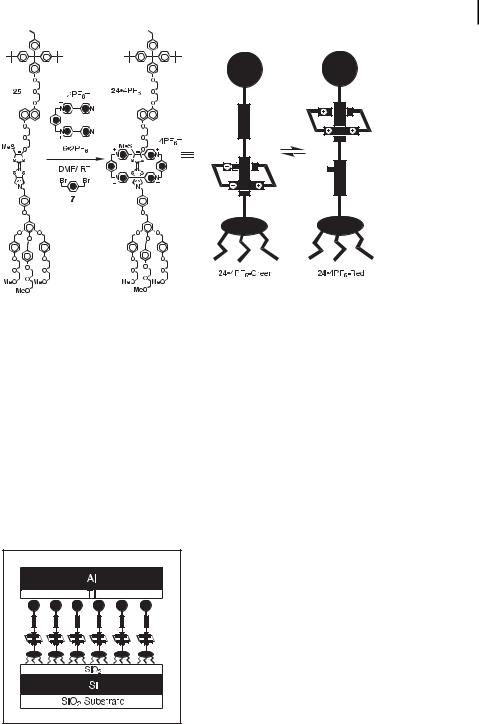
The amphiphilic molecular shuttle 24 4PF6 was synthesized [15a] by using the dumbbellshaped compound 25 as the template for the formation of the encircling tetracationic cyclophane (Figure 17) from its precursor 6 2PF6 and 1,4-bis(bromomethyl)benzene (7). The molecular shuttle 24 4PF6 was isolated as an analytically pure, brown solid after column chromatography using a solution of NH4PF6 in Me2CO as the eluent. Spectroscopic studies indicated the presence of the two stable translational isomers, 24 4PF6-Green and 24 4PF6- Red, in the isolated product. The visible absorption spectrum recorded in Me2CO showed a broad CT band centered at 805 nm, which is characteristic of the co-conformation (24 4PF6- Green) containing the TTF unit located inside the cavity of the tetracationic cyclophane, and a CT band observed as a shoulder at 540 nm, which results from the DNP ring system encircled by the tetracationic cyclophane in the other co-conformation (24 4PF6-Red). The 1H NMR spectrum recorded at 298 K in [D6]acetone also indicated the presence of two coconformational isomers, slowly interconverting on the 1H NMR timescale. The ratio of the two translational isomers was estimated by integration of the two di erent SMe resonances to be approximately 1:1. By means of preparative thin-layer chromatography, it is possible to isolate the translational isomer (24 4PF6-Red), in which the tetracationic cyclophane encircles the DNP ring system. Allowing the solution of the red translational isomer 24 4PF6- Red to stand at room temperature for 24 h results in the formation of the ‘‘original’’ brown solution containing a mixture of the two translational isomers. By employing a first-order kinetic treatment, a rate constant of 2 10 5 s 1 was obtained for this slow shuttling pro-

588 16 Molecular Switches and Machines Using Arene Building Blocks
Fig. 19. The acid/base-controllable switching of the [2]rotaxanes [26 H] 3PF6 and [27 H] 3PF6.
polyether oxygen atoms and the aCH2NH2þCH2 a units in [26 H]3þ and [27 H]3þ. However, upon addition of iPr2NEt to the solutions of [26 H] 3PF6 or [27 H] 3PF6, the dialkylammonium recognition sites are deprotonated and the hydrogen bonds between the crown ether and the dumbbells are destroyed. As a result, the crown ether ring moves to encircle the bipyridinium recognition sites, giving 26 2PF6 and 27 2PF6, respectively. Upon addition of trifluoroacetic acid, protonations restore the dialkylammonium recognition sites and the crown ether ring moves back from the bipyridinium recognition sites to the dialkylammonium recognition sites. The case history outlined in this section has demonstrated how the concept of a molecular shuttle can be transformed into molecular abacus-like switches

16.7 The Evolution of Photochemically Driven Molecular Switches 589
Fig. 20. The photochemically induced dethreading of the pseudorotaxane [4I32]4þ.
that can be both chemically and electrochemically driven. The next section describes how very similar kinds of switches and machines have been designed and operated photochemically in collaboration with the Balzani group and, in the case of the solid-state systems, with the Zink group at UCLA.
16.7
The Evolution of Photochemically Driven Molecular Switches
The [2]pseudorotaxane [4I32]4þ can behave [17a] as a photochemically driven molecular switch (Figure 20) in aqueous solution. When the thread-like compound 32 is added to the tetrachloride salt of the tetracationic cyclophane 44þ, it forms the [2]pseudorotaxane [4I32]4þ in aqueous solution. Next, the deoxygenated solution of the [2]pseudorotaxane [4I32]4þ is irradiated in the presence of an external electron-transfer photosensitizer (P, 9- anthracene-carboxylic acid) and a sacrificial reductant (Red, triethanolamine). It should be noted that the photosensitizer must (i) be able to absorb light e ciently (Process 1 in Figure 20) and (ii) have a su ciently long-lived excited state (P*) so that its irradiation in the presence of the [2]pseudorotaxane [4I32]4þ will lead to the transfer of an electron to one of the bipyridinium units of the cyclophane (Process 2 in Figure 20). The related fast back electron transfer from the reduced cyclophane to the oxidized photosensitizer is prevented by the presence of the sacrificial reductant (Red). As a consequence, the p-p stacking interactions, which hold the 1:1 complex [4I32]4þ together, are seriously impaired, resulting in dethreading of the [2]pseudorotaxane. This process can be monitored by the disappearance of the CT band between the DNP ring system and the cyclophane, and the appearance of the fluorescence of the DNP ring system in the ‘‘free’’ 32. If oxygen is allowed to enter the aqueous solution, the reduced cyclophane 4 3þ is re-oxidized and the [2]pseudorotaxane [4I32]4þ is re-formed.
The same concept has been demonstrated [17b] in the solid state by two di erent approaches, a ording supramolecular machines. In the first system (Figure 21), the supramolecular machines are physically trapped in a rigid, nanoporous, and optically transparent matrix by condensation of a sol-gel silica framework around the [2]pseudorotaxane [4I32]4þ, photosensitizer (P), and sacrificial reductant (Red). The XeCl laser-induced fluorescent emission of the dethreaded DNP ring system in component 32 is monitored. The dethreading process was found to be roughly an order of magnitude slower in the gel than in solution. This reduced rate of dethreading probably results from the stabilization in-



592 16 Molecular Switches and Machines Using Arene Building Blocks
Fig. 24. Photocontrollable molecular machines based on the [2]pseudorotaxanes [8I34]4þ
respectively. Photoirradiation induces intramolecular single electron transfer from the ruthenium metal complex P to an adjacent bipyridinium unit of the cyclophane. As a consequence, the p-p stacking interactions between the cyclophane 354þ and the thread 36 are destroyed. The competing back electron transfer is blocked by the presence of the reductant (Red). Upon introduction of oxygen into the reaction mixture, the bipyridinium unit is oxidized and complexation between the cyclophane 354þ and the thread 36 is restored. The mechanical motions exhibited by this supramolecular machine can be monitored by the intensity of the fluorescence associated with the free DNP-containing thread 36.
Following the evolution of these photochemically driven supramolecular machines, a molecular machine was reported [19] in the form of a molecular-level abacus. It was designed in the shape of the [2]rotaxane 37 6PF6, composed of the p-electron-donating macrocyclic polyether BPP34C10 (1) and a dumbbell-shaped component 38 6PF6, which contains (i) a photoactive Ru(II)-polypyridine complex as one of its stoppers, (ii) a p-terphenyl-type ring system as a rigid spacer, (iii) 4,40-bipyridinium and 3,30-dimethyl-4,40-bipyridinium units as two di erent p-electron-accepting stations, and (iv) a tetraarylmethane group to act as the second inert stopper. The [2]rotaxane 37 6PF6 was synthesized (Figure 25) by a thermodynamically driven slippage reaction between the dumbbell 38 6PF6 and BPP34C10 (1). The mechanical properties of this molecular shuttle were characterized by cyclic voltammetry. The stable translational isomer is the one in which the BPP34C10 component encircles the 4,40- bipyridinium unit, in keeping with the fact that this unit is a much better p-electron acceptor than the 3,30-dimethyl-4,40-bipyridinium unit. Two strategies have been employed in order to