

1Ава VII
ЕРМОКАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ ПЕРЕРАБОТКИ ЕФТЯНЫХ ФРАКЦИЙ
44. Основные представления о катализе и свойствах катализаторов
Большинство химических превращений углеводородов нефти, Цйеющих практическое значение, осуществляется в присутствии |,атализаторов. Катализаторы позволяют снижать энергию акти-”ции химических реакций и тем самым значительно повышать х скорость. В самом общем виде в этом и заключается сущ-ость и значение катализа. Проведение реакции в присутствии |атализаторов позволяет также резко снижать температуру процесса. Для реакций, характеризующихся положительным тепловым эффектом (полимеризация, гидрирование, алкилирование g др.), это имеет особо важное значение, так как высокие температуры с термодинамической точки зрения для них неблагопри" |тны.
;: Любой катализатор активно взаимодействует с исходными реагентами, но его участие в процессе ограничивается только начальными стадиями превращений. В последующих стадиях он Водностью регенерируется и может вновь взаимодействовать с Молекулами реагирующих веществ. Этим и объясняется, что небольшого количества катализатора достаточно для получения очень больших количеств конечного продукта реакции. Факт снижения энергии активации химической реакции за счет образования промежуточных систем с участием катализатора является несомненным. Однако характер самого взаимодействия с катализа-гором может быть самым разнообразным.
I Различают гомогенный и гетерогенный катализ. При гомогенном катализе катализатор и реагирующие вещества образуют однородную систему, например газовые смеси или жидкие растворы. 1ри гетерогенном катализе катализатор чаще всего находится в |ердой фазе, а реагирующие вещества—в газообразном или па-Ообразном состоянии, т. е. в другой фазе. Возможны случаи, дгда реагенты и катализатор—жидкости, но не смешивающиеся Щцежду собойлВ нефтеперерабатывающей промышленности наибо-рее распространены каталитические процессы получения топлив — рсаталитический крекинг, изомеризация, риформйнг, алкилирова-|рие, гидрокрекинг. \
II [Из новых пртщессов на применении катализаторов основано
iee 90 %.j
В последнее время для удобства рассмотрения механизма ка-\ тализа принято все каталитические реакции делить на кислотно" основные и окислительно-восстановительные.
Кислотно-основными, или ионными, называются такие каталитические реакции, которые объясняются присоединением или отщеплением иона водорода (протона), а также реакции, при которых свободная пара электронов у реагирующих веществ или катализатора перемещается без разобщения электронов, образуя координационную связь в комплексном соединении. Отметим здесь же, что согласно теории Льюиса кислотами называются соединения, молекулы которых способны присоединять электронную пару, т. е. включать ее в электронную оболочку одного из своих атомов.
Основаниями называются такие соединения, в молекуле которых имеется неподеленная пара электронов, способная внедряться в электронный слой кислоты. Это наиболее общая трактовка понятий кислоты и оснований. С такой точки зрения в группу кислот помимо обычных кислых веществ попадают и такие соединения, даже не содержащие водорода, как [_А1С1з, РеС1з, ZnCl2, ВРз. Все эти вещества — комплексообразующие катализаторы^
(К кислотно-основным каталитическим реакциям можно отнести каталитический крекинг, полимеризацию, изомеризацию, гидратацию углеводородов и ряд других превращении^
' Окислительно-восстановительными называются такие реакции, которые связаны с переходом электронов, т. е. протекают с разъединением электронной пары при разрыве валентной связи. К ним относятся многочисленные каталитические процессы окисления, гидрирования, дегидрирования и др^Л
Гомогенный катализ. Механизм гомогенного катализа хорошо объясняется теорией промежуточных химических соединений. По этой теории катализатор с реагирующим веществом образует неустойчивое реакционноспособное промежуточное соединение. Энергия активации этого процесса ниже энергии активации некаталитической основной реакции. В дальнейшем промежуточное соединение распадается или реагирует с новой молекулой исходного вещества, освобождая при этом катализатор в неизмененном виде. Эти превращения также характеризуются сравнительно малой энергией активации.
Гетерогенный катализ. При гетерогенном катализе все изменения и превращения веществ происходят на поверхности раздела твердой фазы катализатора и газовой (паровой) фазы реагирующих веществ и в значительной мере связаны с явлением сорбции. Процесс можно расчленить на следующие пять этапов:
1) движение (диффузия) реагирующих молекул к поверхности катализатора;
2) активированная адсорбция (хемосорбция) реагирующих веществ на поверхности катализатора;
3) химическая реакция на поверхности катализатора;
| 4) десорбция (снятие с поверхности катализатора) продуктов Цакции;
^ 5) диффузия продуктов реакции в глубь газовой фазы. Ц Очевидно, скорость всего процесса определяется скоростью са-1вго медленного этапа. Здесь различают два случая. Если диф-1узия исходных и конечных продуктов протекает быстрее, чем JfcMa каталитическая реакция, то скорость процесса целиком за-Йсит от состава и свойств поверхности катализатора. В этом йучае говорят, что процесс протекает в кинетической области. аоборот, если диффузия происходит медленнее, чем завершаются \ превращения на поверхности катализатора, то общая ско-сть реакции будет определяться скоростью диффузии. В этом ^чае говорят, что процесс протекает в диффузионной области. йижение энергии активации в гетерогенных каталитических ре-вдиях достигается в результате сложных физико-химических 1юцессов, характеризующих стадию активированной адсорбции |емосорбции).
| Хемосорбция — это образование достаточно прочного мономо-|кулярного слоя реагирующих веществ на поверхности катализа-&ра. Хемосорбция протекает за счет валентных сил катализато-||, по сути близка к настоящей химической реакции и поэтому риводит к значительному ослаблению связей в реагирующих мо-^кулах.
, Активированная адсорбция молекул реагирующих веществ роисходит не на всей свободной поверхности твердого катализа-'ЩЬра, а только на так называемых активных центрах, где запас Цюбодной энергии больше. Это могут быть острые углы, пики, азличные неровности, ребра кристаллов, химически неоднород-ые участки и т. д. В целом, чем сильнее развита общая Оверхность, тем больше на ней активных центров. Поэтому эвышение активности катализатора часто связано с его высоки степенью измельчения и хорошо развитой пористой струк-1рой.
^ Таким образом, общий принцип действия гетерогенных ката-Взаторов заключается в том, что молекулы реагирующих веществ, ёалкиваясь с активными центрами катализатора, могут образо-tTb различные нестойкие промежуточные соединения (радикалы йи ионы). От реакционной способности и природы этих соеди-|аий зависят и скорость протекающих реакций, и состав конеч-Цх продуктов.
KB практике подбора и применения гетерогенных катализаторов ||обходимо учитывать их' селективность, активность и стабиль-Цеть, определяющую срок службы.
Ц Под" селективностью, или избирательностью, катализатора поймают его способность ускорять только одну или несколько хи-|Вяеских реакций определенного типа из числа термодинамически йроятных в данных условиях для данного сырья. Это весьма |1жное свойство катализатора связано как с его составом, так и ^.условиями применения,
Активность катализатора характеризует его производительность. Чем активнее катализатор, тем меньше его нужно для превращения определенного количества исходных веществ в конечные продукты за единицу времени. 'Активность твердого катализатора зависит главным образом от состояния его поверхности. Катализаторы обычно применяются в виде таблеток, шариков или зерен небольших размеров. Для увеличения площади поверхности часто катализатор наносят на подложку (носитель), обладающую пористой поверхностью. В качестве носителей применяют активный уголь, пемзу, кизельгур, оксид алюминия, силикагель и искусственные цеолиты различных марок. Носитель повышает активность катализатора, придает ему механическую прочность и уменьшает его расход^
',_Активность многих катализаторов удается повысить добавлением небольшого количества так называемых промоторов, или йкгивагороб_зДействие активаторов может быть различным. Одни вещества увеличивают внутреннюю поверхность катализатора, т. е. воздействуют на его структуру и способствуют ее сохранению во время работы. Такие промоторы получили название структурных. Другие активаторы изменяют химический состав поверхности катализатора, увеличивают число активных центров. Такие активаторы получили название химических.
Активность катализаторов обычно определяют на лабораторных установках, где осуществляется тот технологический процесс, для которого предназначен данный катализатор. Активность оценивают по выходу целевого продукта или степени превращения исходного сырья. Иногда испытуемый образец сравнивают с эталонным, активность которого известна. Активность катализаторов каталитического крекинга характеризуется так называемым индексом активности — выходом (в % от сырья) фракции до 200 °С, полученной в результате каталитического крекинга эталонного сырья в стандартных условиях на лабораторной установке.
; Катализатор после каждого элементарного акта реакции остается неизменным. Поэтому, казалось бы, он должен всегда работать неограниченно долго. Однако на практике срок службы катализатора всегда конечен, так как активность его со временем падаетД
^Спад активности катализатора (дезактивация) часто называется его утомлением или старенивм.^Прпчинъ} этого явления различны. Чаще всего снижение активности катализатора наступает в результате отложения на его активных центрах продуктов реакции (кокса, смолы, парафинов и т. д.) или вследствие воздействия некоторых примесей в сырье, называемых каталитическими ядами^] Последние особенно опасны, так как достаточно их ничтожного количества для полной дезактивации катализатора. Это заставляет весьма внимательно относиться к составу сырья. Часто для предотвращения отравления катализатора приходится исходное сЫрье подвергать дополнительной очистке. Действие катали-
деских ядов объясняется их необратимой адсорбцией на актив-й центрах катализатора.
;;!- Наиболее чувствительны к ядам металлические катализаторы, йдержащие Fe, Co, Ni, Pd, Ir, Pt, Cu, Ag. Часто встречаются еле-Юющие каталитические яды: сероводород и другие серусодер-|ащие соединения, оксид углерода, азотистые основания, галоге. йд, соединения фосфора, мышьяка и сурьмы, а также металлы JNi, Fe, V), содержащиеся в тяжелом нефтяном сырье.
|. К причинам дезактивации катализаторов относятся также из-^нение структуры его поверхности, спекание и другие процессы, вторые приводят к уменьшению поверхности или числа активных ентров и могут быть вызваны условиями данного технологиче-[toro процесса, главным образом температурой.
^ Восстановление активности катализатора называется регенерацией. Способы регенерации различны. Углистые и смолистые Тложения выжигают с поверхности катализатора в токе воздуха, Окисленные катализаторы восстанавливают действием водорода ри определенной температуре. Катализаторы, содержащие ред* |йе элементы (например, торий), регенерируют химическим пу* |ём. Редкий элемент извлекают из отработанного катализатора и |вовь используют для приготовления свежих порций этого же Катализатор^
iB качестве катализаторов применяются разнообразные веще” гва: восстановленные металлы, оксиды, кислоты, основания, со* и, сульфиды металлов, некоторые органические соединения, Под-ор катализаторов, изучение условий их изготовления и примене' ия, пути регенерации и другие вопросы, связанные с катали* ... эм,—все это в настоящее время неотъемлемая часть нефтехими” Щйеской технологии,!
U. КАТАЛИЗАТОРЫ И МЕХАНИЗМ КАТАЛИТИЧЕСКОГО КРЕКИНГА
Ц' Крекинг нефтяного сырья в присутствии катализаторов, или, 'Коротко, каталитический крекинг — в настоящее время один из Основных методов производства базовых компонентов автомобильных бензинов. Применение катализатора в крекинг-процессе вно-Цит значительные изменения как в механизм протекающих превращений углеводородов, так и в состав получаемых продуктов. Преимущества каталитического крекинга заключаются, во-первых, | том, что в результате общего ускорения процесса удается не-|колько снизить температуру крекинга и проводить процесс при |йзком давлении; во-вторых, и это главное, селективное действие Цвтализатора ускоряет такие реакции, которые приводят к накоп-|мению в крекинг-бензине аренов, изоалканов и изоалкенов, обла-Цйющих большими октановыми числами.
I; Процесс проводят в паровой фазе при 450—525 °С под давлением не выше 0,15 МПа в присутствии алюмосиликатного катализатора,
До недавнего времени на отечественных заводах применялиЗ аморфные природные синтетические алюмосиликаты, содержащИ в среднем 10—25 % А^Оз, 80—75 % SiOz и некоторое количеств воды, а в виде примесей—оксиды железа, магния, кальция] натрия. Эти катализаторы обладают индексом активности поря! ка 35—38. ;
В настоящее время почти все установки каталитического кр| кинга переведены на работу с синтетическими кристаллическикЯ алюмосиликатными катализаторами, содержащими в своем сост^ ве цеолиты. %
Цеолитами называются природные и искусственные алюмосв ликаты, обладающие чрезвычайно тонкопористой структурой. Он представляют собой водные алюмосиликаты натрия, кальция ' других металлов, т. е. это соли алюмокремниевых кислот: напр^ мер, цеолит под индексом 13Х — Nage [(AK^se (ЗЮ2)юб] •267Н2Ё Все цеолиты имеют кристаллическое строение с четко вв раженной структурой. Их общее свойство — наличие мельча! ших полостей, сообщающихся между собой через “окна” (порыЩ Число полостей в кристалле цеолита очень велико. Их общи^ объем может достигать половины всего объема кристалла. Имен| но благодаря этому обезвоженные цеолиты являются прекрасный ми адсорбентами. Но в отличие от других адсорбентов (активы! уголь, силикагель, оксид алюминия и др.) размер пор у цеолито! очень мал и все поры имеют одинаковый диаметр. Для разный типов синтетических цеолитов размер пор колеблется от 0,3 да 1,3 нм. Следовательно, поры цеолита имеют молекулярные ра| меры, благодаря чему цеолиты обладают как бы просеивающими свойствами, т. е. могут пропускать в полости кристалла и адсо| бировать- молекулы только определенного размера, не превышаю щие диаметра пор данного цеолита. Поэтому они получили такж название молекулярных сит.
Синтетические цеолиты выпускаются трех типов: А, X, Y. Цес литы типа А имеют поры 0,3—0,5 нм и применяются для осушк|| и очистки углеводородных газов от углекислого газа и серовод(| рода, а также для адсорбции и отделения нормальных алкансй из их смеси с углеводородами других классов. Цеолиты типа 3 имеют размер пор 0,8—1,3 нм, а типа Y—0,8—0,9 нм. Цеолита Х и Y нашли большое применение в производстве катализаторов
Катализаторы на основе цеолитов 10Х и 10Y (шариковые-” цеокар-2, АШНЦ-3, микросферический РСГ-2ц и другие) харавйЦ теризуются индексом активности близким к 50. Некоторые из эти?" катализаторов содержат оксид рения, что благотворно сказывав! ся на селективности действия катализатора и позволяет получат большие выходы бензина.
Содержание самих цеолитов в цеолитсодержащих катализатс pax невелико (3—15 %). В процессе приготовления катализатор! модифицированные металлами (хромом, рением, платиной, палладием и др.) цеолиты вводят в матрицу (подкладку) неорганич^ ских оксидов <SiOz, А1г0з, глины и др.).
Состав,
%
SiOa
AlsOa РеаОз
NaaO
84,3
13,0 0,13 0,88
кг/м3
Индекс активности 48
Индекс стабильности 51,5
Особенностью катализаторов каталитического крекинга является их быстрая дезактивация. Поры катализатора закоксовыва-ются через 10—15 мин работы. Поэтому необходимо крекинг все время чередовать с регенерацией катализатора. Каталитические процессы с частой сменой циклов работы и регенерации получили название цикличных. Регенерация заключается в выжигании кокса и смолистых отложений с поверхности катализатора воздухом при 540—680 °С. Для предохранения катализатора от местных перегревов воздух разбавляют инертными газами. Однако число циклов регенерации не беспредельно.
Свойство катализатора сохранять активность во время эксплуатации называется стабильностью активности. С течением времени вследствие постепенного уменьшения удельной активной поверхности катализатора и изменения его пористой структуры индекс активности катализатора падает. Это происходит в результате механического истирания частиц катализатора, спекания его и постепенного отравления активных центров металлами, содержащимися в сырье в виде сложных металлорганических соединений типа пор-фириновых комплексов, а также продуктов коррозии. Частичная замена катализатора проводится периодически или непрерывно.
Катализаторы каталитического крекинга должны также обладать механической прочностью, устойчивостью к истиранию, к действию высокой температуры, водяного пара и к резким изменениям температур.
Изучение строения и свойств алюмосиликатного катализатора показало, что он обладает явно выраженной кислотностью. В наибольшей степени это относится к цеолитсодержащим катализаторам. Смеси безводных оксидов кремния и алюминия мало активны. Следовательно, катализаторами следует считать только гидра-тированные алюмосиликаты. Отметим, что вода удерживается в катализаторе даже при нагревании его до 700 °С. Носителем активности поверхностного слоя катализатора считается соединение (HAlSi04)x, которое может диссоциировать на ион водорода (протон) и ион AlSiOi. Катализатор способен к водородному обмену, в этом и проявляется его активность.
Наиболее распространенной теорией, объясняющей механизм каталитического крекинга на алюмосиликатных .катализаторах, в настоящее время является теория карбоний-иона. В основе ее лежат следующие представления.
Ионы карбония аналогичны ионам аммония и содержат трехвалентный положительно заряженный атом углерода. Легче всего они образуются при взаимодействии протона катализатора
с олефинамш
R - СН = СН, + Н+—>R - СН - СНз
Участие в реакции водорода катализатора доказано опытами с мечеными атомами (дейтерием). Ионы карбония образуются в хемосорбированном слое молекул и являются активными промежуточными продуктами многих реакций, протекающих при каталитическом крекинге.
Путем внутренней перегруппировки или взаимодействия с другими молекулами неустойчивые ионы карбония стремятся перейти в более устойчивые формы. Они отличаются очень большой реакционной способностью и могут вступать в самые разнообразные реакции.
Для ионов карбония наиболее характерны следующие превращения.
1. Перегруппировка атомов в молекуле с перемещением водо-' рода или метальных групп, т. е., иначе говоря, скелетная изомеризация, приводящая к образованию изомерного иона:
Р—СН—СНа—СНз
СНз
R—C—СНз
+
2. Взаимодействие с нейтральными молекулами с образованием новых ионов карбония и новых молекул как предельных, так и непредельных:
R—CH—СНз
+ С4Ню R—CH—СНз + С4Нд
С<Н^
+ Р—СНг—СНз .C4H?+R—CH==CH2
3. Распад карбоний-иона с большим числом углеродных атомов, как правило, происходит по р-связи, считая от заряженного атома углерода. Продукты распада — алкен и карбоний-ион с меньшим числом углеродных атомов:
СНз—СН—СНг—Р —> СНз—СН^СНа+Р4-
+
В целом, согласно теории ионов карбония, механизм каталитического крекинга представляется в виде цепных реакций. Зарождение цепи происходит под действием иона водорода катализатора:
НА —> Н^А" где НА — условное изображение алюмосиликатного катализатора в виде кислоты.
Развитие цепи идет по линии разнообразных превращений Конов карбония и водородного обмена с нейтральными молекула-[дй. Наконец, обрыв цепи происходит в результате реакции между Ионом карбония (катион) и анионом алюмосиликата:
R—CH- -СНз + А' R—CH^CHs + НА
Катализатор в итоге регенерируется, и процесс может начинаться снова, если поверхность катализатора еще сохраняет ак-
1йвность.
1. Характерная черта каталитического крекинга — развитие про-Дессов перераспределения, или диспропорционирования, водорода. |'В общем виде этот процесс заключается в том, что молекулы, "адсорбированные на катализаторе, склонны терять водород, т. е. дегидрироваться. Это приводит к увеличению ненасыщенности &, как следствие, к более прочной адсорбции на катализаторе. В итоге вновь образовавшиеся непредельные углеводороды начинают полимеризоваться и превращаться в конечный продукт процесса — кокс, который необратимо откладывается на катализаторе. Постепенно все активные центры закрываются коксом, |й катализатор необходимо регенерировать.
I;, Итак, мы видим, что при каталитическом крекинге кокс — это Цйродукт основного каталитического процесса, и его накопление на 1'Катализаторе неизбежно. При термическом крекинге кокс накап-Цяивается в результате вторичных химических превращений и яв-|;ляется продуктом высокотемпературных побочных реакций кон-|;денсации, роль которых может быть снижена путем изменения
^Параметров процесса.
Ц^ Другое следствие развития процесса диспропорционирования Цйодорода — более насыщенный характер жидких и газообразных ^продуктов крекинга, чем- при термическом крекинге. Водород, те-|ряемый реагирующими молекулами, адсорбированными на катализаторе, расходуется на насыщение различных осколков молекул, |й прежде всего на гидрирование изомеризованных алкенов. Следовательно, одни молекулы углеводородов теряют водород, а другие Цэа их счет насыщаются водородом. В этом и заключается процесс Ййспропорционирования водорода.
Остановимся кратко на поведении при каталитическом крекин-; углеводородов различных гомологических рядов.
Алканы. Так же как и при термическом крекинге, алканы рас-аадаются на алкен и алкан меньшей молекулярной массы. Распад Происходит в нескольких местах углеродной цепи, но не на самом |е конце. Выходы метана, этана и этилена незначительны. В газе Скапливаются углеводороды Сз—С4. Скорость распада в десятки Ййз больше, чем при термическом крекинге.
|^' Алканы не попадают в хемосорбированный слой. Но и за пределами этого слоя проявляется поверхностная энергия катализатора. В результате в непосредственной близости от поверхности Катализатора создается зона повышенной концентрации углеводо-
родных молекул с ослабленными связями С—С. Это и снижает энергию активации реакций распада. При каталитическом крекинге отмечена также возможность дегидрирования углеводородов с числом углеродных атомов в молекуле более четырех. Образующиеся при этом высшие алкены в свою очередь распадаются.
Алкены. Скорость распада алкенов при каталитическом крекинге в тысячи раз больше, чем при термическом крекинге. Причины этого .и механизм превращений уже описаны выше. Помимо распада алкены вступают в реакции полимеризации — деполимеризации, перераспределения водорода, изомеризации, циклизации.
Особенно разнообразны реакции изомеризации. Здесь имеют место структурная перегруппировка, перемещение двойной связи вдоль цепи, возможна и цис-, транс-изомерия.
Циклоалканы. Реакции, характерные для крекинга циклоалка-нов — деалкилирование, дегидрирование, распад кольца,— ускоряются в присутствии катализатора в 500—4000 раз. 3 отличие от термического крекинга, кроме того, заметно развиты перечисленные ниже реакции:
распад кольца с образованием изоалкенов
(—/—СНз изомеризация циклов
СНз—СН2—СН==СН—С—СНз СНз
___/СНз
перераспределение боковых цепей (миграция заместителей) -"СНаСНз ^-^ss^ ^'СНз ^"•"s^ ^СНз
^^03
![]()

Дегидрирование и деалкилирование циклоалканов протекает с перераспределением водорода. В результате в продуктах крекинга накапливаются арены и алканы. Например:
^СНаСНдСНз /^^
![]()
Бициклические циклоалканы в результате дециклизации, дегидрирования, деалкилирования и изомеризации дают начало моноциклическим аренам, циклогексанам, циклопентанам и ал-канам.
Для всех реакций распада циклоалканов, так же как и для углеводородов с открытой цепью, характерно образование осколков не ниже Сз.
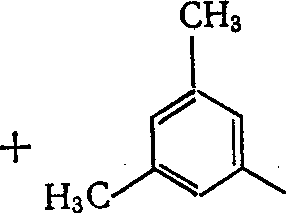
|Арены. Скорость и направление превращений аренов при ката-•Йическом крекинге в большой мере зависит от строения и моле-аярной массы крекируемого углеводорода. ЙГомологи бензола преимущественно полностью теряют боковые Йи, что приводит к накоплению бензола. Труднее всего креки-жтся толуол. По мере увеличения длины боковой цепи и ее раз-Увления глубина деалкилирования резко возрастает. |Полиметилированные гомологи бензола легко изомеризуются Перераспределением метильных групп. Так, при крекинге п-кси-|ла в, продуктах реакции обнаружены толуол, триметилбензол,
уи о-ксилолы:
СНз
СНз
СНз
СНз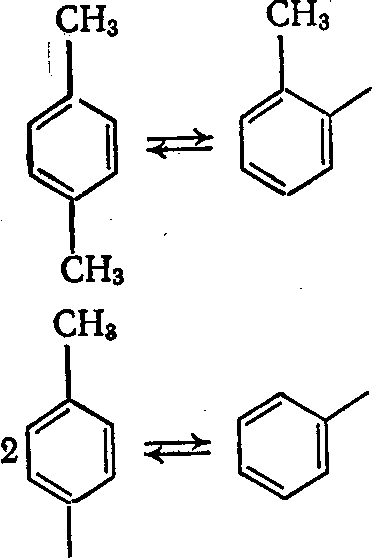

амы. 1 Конденсированные полициклические углеводороды очень легко
Цеорбируются на катализаторе и крекируются на нем до кокса. |&нзол практически в реакции не вступает и поэтому накапливайся в крекинг-бензине.
У Особенности механизма каталитического крекинга в сравнении (.термическим крекингом резко сказываются на составе и свой-Йвах газа и бензина. В газе меньше низкомолекулярных композитов, но гораздо больше изобутана. Бензины обогащаются
а&алканами и аренами.
f
Щ6. СЫРЬЕ, ПАРАМЕТРЫ И ПРОДУКТЫ КАТАЛИТИЧЕСКОГО КРЕКИНГА
|| В процессе каталитического крекинга целевым продуктом яв-Цйтся бензиновая фракция с высоким октановым числом. Кроме днзина в этом процессе получаются еще углеводородный газ, Цгкий газойль (фракция 195—350 °С), тяжелый газойль и кокс. ^окс, как уже упоминалось выше, откладывается на катализаторе Цсжигается при регенерации катализатора. Количество и качест-получаемых продуктов, а также количество образующегося
кокса зависят как от качества сырья, так и от параметров процесса.
Сырье. Основными видами сырья для каталитического крекинга являются фракции, выкипающие, как правило, в пределах 200—500 °С. К ним относятся керосино-газойлевые фракции (200—350 °С), вакуумный газойль прямой гонки и продукты вторичных процессов: газойли коксования, термического крекинга и гидрокрекинга. Более легкое сырье применяют для получения компонента базового авиационного бензина, более тяжелое — автобензина. Применяют также смешанное сырье — как по фракционному составу (225—490 °С, 265—500 °С), так и по происхождению. Смешанное сырье используют для производства автобензина.
Качество исходного сырья оказыва-ет большое влияние на работу катализаторов. Повышение содержания в сырье непредельных и полициклических аренов или утяжеление фракционного состава ведет к повышенному коксообразованию и быстрой дезактивации катализатора. По этой причине газойли коксования и термического крекинга перерабатывать сложнее, чем прямогонное сырье. Наличие в сырье смол, серу- и азотсодержащих соединений и металлов также снижает активность и селективность катализатора. Поэтому иногда при крекинге даже прямогонных газойлей, полученных из смолистых и сернистых нефтей, приходится ограничивать конец кипения сырья на уровне 480—490 °С. Ниже в качестве примера приведены свойства вакуумного газойля ромаш-кинской нефти — характерного сырья каталитического крекинга;
Плотность pi° 0,9165-0,9231
Фракционный состав
температура начала кипения, °С 345—352 10% перегоняется при температуре, °С 397—403 50% перегоняется при температуре, °С 444—456
Содержание, %
серы 1,79—1,88 азота 0,11—0,07 никеля (3—4) • Ю-8 ванадия 1 • Ю-3 Коксуемость, % , 0,39—0,43
Хорошие результаты крекинга достигаются на предварительно гидроочищенном сырье. При гидроочистке значительно снижаются содержание серы, азота и металлов в сырье, а также его коксуемость. Крекинг подготовленного сырья приводит к уменьшению выхода кокса и газа и увеличению выхода бензина. Улучшается качество полученных продуктов: повышается октановое число бензина, содержание серы в жидких продуктах крекинга снижается настолько, что эти продукты не нуждаются в последующей очистке от серы. Положительный эффект дает также предварительное удаление смолисто-асфальтеновых веществ из сырья методом де-асфальтизации бензином или сжиженным пропаном.
Параметры процесса. Основными параметрами каталитического крекинга являются температура, время контакта паров сырья