


120 |
Chapter 4 |
8.ENZYME KINETICS AND MECHANISM
Application of CBS extrapolations to the  isomerasecatalyzed conversion of
isomerasecatalyzed conversion of  to the
to the  isomer (Fig. 4.10) provides a test case for extensions to enzyme kinetics. This task requires integration of CBS extrapolations into multilayer ONIOM calculations [56, 57] of the steroid and the active site combined with a polarizable continuum model (PCM) treatment of bulk dielectric effects [58-60]. The goal is to reliably predict absolute rates of enzyme-catalyzed reactions within an order of magnitude, in order to verify or disprove a proposed mechanism.
isomer (Fig. 4.10) provides a test case for extensions to enzyme kinetics. This task requires integration of CBS extrapolations into multilayer ONIOM calculations [56, 57] of the steroid and the active site combined with a polarizable continuum model (PCM) treatment of bulk dielectric effects [58-60]. The goal is to reliably predict absolute rates of enzyme-catalyzed reactions within an order of magnitude, in order to verify or disprove a proposed mechanism.
Deuterium substitution for the migrating  proton demonstrates that the enzyme transfers it by a stereospecific intramolecular path
proton demonstrates that the enzyme transfers it by a stereospecific intramolecular path

Complete Basis Set Models |
121 |
to the 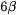 position without solvent exchange [61-63]. The structure of the active site (Fig. 4.11) provides a ready explanation for the observed stereochemistry. The
position without solvent exchange [61-63]. The structure of the active site (Fig. 4.11) provides a ready explanation for the observed stereochemistry. The 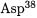 (aspartate residue 38) is well situated to escort the migrating proton, while
(aspartate residue 38) is well situated to escort the migrating proton, while  (aspartic residue 99) and the
(aspartic residue 99) and the  (tyrosine residue 14) stabilize the dienolate intermediate through hydrogen bonding [64].
(tyrosine residue 14) stabilize the dienolate intermediate through hydrogen bonding [64].
The accepted mechanism is rather complicated [64]. First the  proton of the substrate transfers to
proton of the substrate transfers to  then the (now protonated) carboxyl group of
then the (now protonated) carboxyl group of  rotates about the
rotates about the  bond to position the migrating proton over carbon 6 of the substrate, and finally the migrating proton transfers to the
bond to position the migrating proton over carbon 6 of the substrate, and finally the migrating proton transfers to the  position on the
position on the 
 product. The seven stationary points on the potential energy surface are therefore the reactant, two intermediates, the product, and three transition states (Fig. 4.12). Before we can calculate energies and reaction rates, we must first locate these structures on the potential energy surface, and then determine by a frequency calculation whether each stationary point is a local minimum, a first-order saddle point, or a structure that is not relevant to the reaction mechanism. These preliminary structure calculations verify whether the proposed mecha-
product. The seven stationary points on the potential energy surface are therefore the reactant, two intermediates, the product, and three transition states (Fig. 4.12). Before we can calculate energies and reaction rates, we must first locate these structures on the potential energy surface, and then determine by a frequency calculation whether each stationary point is a local minimum, a first-order saddle point, or a structure that is not relevant to the reaction mechanism. These preliminary structure calculations verify whether the proposed mecha-

122 |
Chapter 4 |
nism is qualitatively consistent with the potential energy surface for our quantum-mechanical model problem.
The key functional groups for the active site are widely separated in the amino acid sequence (14, 38, and 99), as is frequently the case. We therefore elected to omit the intervening residues from our calculations entirely, and instead rely on optimization of the transition state structures to position these functional groups.
The three levels of theory selected for the preliminary ONIOM calculation [57] were: CBS-4M, HF/3-21G(*), and MM/UFF [65, 66]. Note that HF/3-21G(*) is the level of theory employed in the geometry and frequency calculations for the CBS-4M//HF/3-21G(*) compound model. Thus, the first task was the optimization of the structures for the seven stationary points on the potential energy surface using a two-level ONIOM calculation employing HF/3-21G(*) for both the high-level and the medium-level regions from the planned CBS ONIOM single-point calculations. The partition of the enzyme-substrate complex into high-, medium-, and low-level regions is indicated in Fig. 4.13. The first transition state structure was easily found with the QST2 procedure of Schlegel and co-workers [67]. However, the others proved more problematic.
The optimum structure for the first transition state placed the  residue (i.e. our formic acid) reasonably close to the position in which it is found in the enzyme-inhibitor crystal structure [64]. However, this functional group is not at all rigid in our model problem. It
residue (i.e. our formic acid) reasonably close to the position in which it is found in the enzyme-inhibitor crystal structure [64]. However, this functional group is not at all rigid in our model problem. It

Complete Basis Set Models |
123 |
shifts toward the 3-carbonyl in the fully optimized reactant and shifts in the opposite direction as we proceed to the product. We therefore constrained the CH hydrogen of this formic acid to be equidistant from carbons 4 and 6 of the androstene at the optimum distance for the first transition state. This emulated a rigid enzyme active site equally adept at catalyzing both proton transfer reactions. No other constraints were used in the optimization of the seven stationary points of the potential energy surface (Fig. 4.12). The 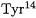 and
and  residues, and the substrate were completely free to move and adjust their geometries as the reaction proceeded.
residues, and the substrate were completely free to move and adjust their geometries as the reaction proceeded.
The optimized structures for the reactant, the first transition state, and the first enolate intermediate (Fig. 4.14) illustrate the progress along the reaction path and the nature of the intermediate (Table 4.9). The bond between the  hydrogen and carbon 4 lengthens from 1.122 Å to 1.903 Å as the bond order decreases from 0.853 to 0.065. Meanwhile, the distance from this hydrogen to the oxygen of
hydrogen and carbon 4 lengthens from 1.122 Å to 1.903 Å as the bond order decreases from 0.853 to 0.065. Meanwhile, the distance from this hydrogen to the oxygen of 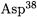 decreases from 1.794 Å to 0.991 Å and the bond order increases from 0.370 to 0.935 as the O–H bond of the intermediate is formed. The O–H bond length of the carboxylic acid from
decreases from 1.794 Å to 0.991 Å and the bond order increases from 0.370 to 0.935 as the O–H bond of the intermediate is formed. The O–H bond length of the carboxylic acid from  increases from 0.999 Å to 1.082 Å as the bond order decreases from 0.847 to 0.636. At the same time the distance from this hydrogen to the C-3 carbonyl oxygen decreases from 1.617 Å to 1.364 Å as this bond order increases from 0.292 to 0.489. The intermediate would seem to be best described as a partially protonated
increases from 0.999 Å to 1.082 Å as the bond order decreases from 0.847 to 0.636. At the same time the distance from this hydrogen to the C-3 carbonyl oxygen decreases from 1.617 Å to 1.364 Å as this bond order increases from 0.292 to 0.489. The intermediate would seem to be best described as a partially protonated

124 |
Chapter 4 |
enolate ion. The negative charge that was localized on the carboxylate of  in the reactant has been delocalized over the dienolate and
in the reactant has been delocalized over the dienolate and  in the intermediate.
in the intermediate.
We performed CBS-4M single point energy calculations at these stationary points. The barrier height for the first proton transfer and the relative energy of the first dienolate are quite sensitive to the level of theory and basis set employed (Table 4.10 and Fig. 4.14). The initial  (i.e. formate ion) carries the full negative charge in
(i.e. formate ion) carries the full negative charge in

Complete Basis Set Models |
125 |
our model and is thus more sensitive to the Hartree-Fock basis set.
Hence, the |
calculated Hartree-Fock barrier increases as the basis set |
is improved. |
The correlation energy favors the transition state, as is |
generally the case. The effects on the enolate are intermediate. The modest changes from the CBS extrapolations suggest convergence to within 1 or 2 kcal/mol. Note that the total barrier height is the sum of the Hartree-Fock and correlation energy contributions, e.g. the MP2/6- 31+G(d’,p’) barrier height is the sum of 14.05 kcal/mol from the HartreeFock component and -10.23 kcal/mol from the second-order component, giving a total of 3.82 kcal/mol for the MP2 barrier.
Refinements of these calculations would include a CBS-QB3 study of the dienolate and  portion of our model. Since we have only used a two-layer ONIOM calculation for the geometry optimizations, a refined geometry optimization with a B3LYP/6-311G(d,p) treatment of this region would be straightforward. We should also use the IRCMax procedure [40] to determine the CBS transition state geometry and energy. Such refinements should be a part of any quantitative CBS study of enzyme kinetics. Nevertheless, the preliminary results listed in Table 4.10 clearly demonstrate the importance of achieving basis set convergence for both the SCF and the correlation energies. Even when the electronic structure is as simple as in these proton transfer reactions that maintain a closed-shell structure throughout the reaction path, low levels of theory can be quite misleading.
portion of our model. Since we have only used a two-layer ONIOM calculation for the geometry optimizations, a refined geometry optimization with a B3LYP/6-311G(d,p) treatment of this region would be straightforward. We should also use the IRCMax procedure [40] to determine the CBS transition state geometry and energy. Such refinements should be a part of any quantitative CBS study of enzyme kinetics. Nevertheless, the preliminary results listed in Table 4.10 clearly demonstrate the importance of achieving basis set convergence for both the SCF and the correlation energies. Even when the electronic structure is as simple as in these proton transfer reactions that maintain a closed-shell structure throughout the reaction path, low levels of theory can be quite misleading.
Calculations of bulk dielectric effects using the polarizable continuum model of Tomasi and co-workers [58-60] gave an increase in the barrier height to 12.5 kcal/mol with the recommended [68] dielectric
constant |
equal to 18. This is the direction of change |
one would |
expect. |
The dielectric medium stabilizes the charge of the |
residue |
(which is completely exposed on the surface of our model) more than the charge of the enolate (which is enclosed in the interior of our model). The calculated barrier height is now in a good agreement with the experimental rate constant,  [64, 69], which implies a Gibbs free energy of activation
[64, 69], which implies a Gibbs free energy of activation 
However, such a large effect raises questions about the accuracy of these PCM corrections. Within the context of high-accuracy methods such as the CBS models, it would be prudent to interpret bulk dielectric effects larger than 1 or 2 kcal/mol as an indication that our model does not
include sufficient detail in the region of the |
residue. We conclude |
|
that a definitive computational study of the |
isomerase |
|
should include the entire |
residue and probably some explicit water |
|
molecules in this region as well.
126 |
Chapter 4 |
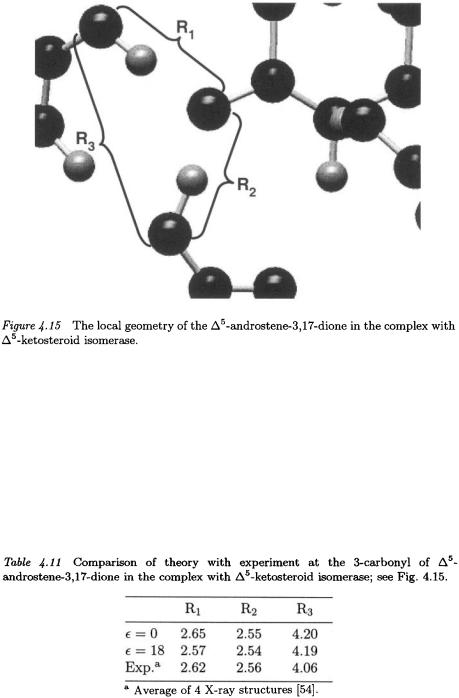
To close on a more positive note, we observe that the computed geometry of the enzyme-dienolate complex in the vicinity of the 3- carbonyl is insensitive to the assumed dielectric constant and is in close agreement with X-ray structures of enzyme-inhibitor complexes (see Table 4.11 and Fig. 4.15). It is really quite remarkable that 4 billion years of random walk by mother nature and a few hours of optimization with a quantum chemistry program such as Gaussian™ (starting with the correct functional groups) lead to the same structure for the active