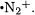Theoretical Thermochemistry of Radicals |
167 |
3.GEOMETRIES
The accurate determination of thermochemical properties can depend greatly on the quality of the optimized geometry. It is therefore necessary to assess the performance of various procedures for obtaining reliable radical geometries. Tables 6.2 and 6.3 present bond lengths for a selection of radicals [22] optimized at several commonly-used levels of theory and compared with experiment [26, 27]. Also included are mean absolute deviations (MADs), mean deviations (MDs), and largest deviations (LDs) from experiment. A positive sign for an MD or LD indicates an overestimation by a given level of theory.
As can be seen from the mean absolute deviations from experiment, all levels of theory give good overall performance for bond lengths. The poorest result for most of the theoretical procedures is observed for the O–Cl bond length in  which is overestimated (by 0.023 - 0.061 Å) by all the methods listed in Tables 6.2 and 6.3. This appears to be a consequence of basis set deficiencies, with improved geometries being obtained at all levels of theory with larger basis sets [28]. The
which is overestimated (by 0.023 - 0.061 Å) by all the methods listed in Tables 6.2 and 6.3. This appears to be a consequence of basis set deficiencies, with improved geometries being obtained at all levels of theory with larger basis sets [28]. The  radical has therefore been excluded from the statistical analysis of the results.
radical has therefore been excluded from the statistical analysis of the results.
The URCCSD(T)/cc-pVTZ level of theory performs the best (Table 6.2), with an MAD of 0.006 Å and an LD of only  The positive mean deviation from experiment (+0.004 Å) indicates that this method slightly overestimates most bond lengths. This has also been previously noted by Martin [29].
The positive mean deviation from experiment (+0.004 Å) indicates that this method slightly overestimates most bond lengths. This has also been previously noted by Martin [29].
The DFT-based, computationally inexpensive UB3LYP/6-31G(d) and UB3LYP/cc-pVTZ methods also perform quite well, with MADs of 0.008 Å and 0.007 Å, respectively (Table 6.3). The LDs (-0.024 and -0.031 Å, respectively) for these methods are larger in magnitude than that of URCCSD(T). Martin et al. [30] found for a small set of closed-shell molecules a significant improvement in bond lengths at the UB3LYP level of theory upon going from the cc-pVDZ to the cc-pVTZ basis set. However, for the radicals of Table 6.3, there is very little difference in the performance of the UB3LYP/6-31G(d) and UB3LYP/ccpVTZ approaches. Bond lengths are slightly overestimated with the 6-31G(d) basis set (MD of +0.005 Å) and slightly underestimated with the cc-pVTZ set (MD of -0.003 Å).
The UQCISD/6-31G(d) level of theory performs marginally less well than its UB3LYP counterpart, with an MAD of 0.012 Å and an LD of +0.030 Å, while slightly overestimating (MD of +0.012 Å) most bond lengths.

168 |
Chapter 6 |
As expected, the UMP2(fu) and RMP2 methods (overall MADs of 0.016 Å and 0.015 Å, respectively) give very similar results for species with minimal spin-contamination. For species displaying significant spincontamination ( and
and  ), the UMP2 approach generally yields significantly shorter bond lengths than experiment while the RMP2 method often significantly overestimates them, in agreement with previous observations [31]. Large deviations from experiment (> 0.030 Å) are also observed at the UMP2 and RMP2 levels of theory for
), the UMP2 approach generally yields significantly shorter bond lengths than experiment while the RMP2 method often significantly overestimates them, in agreement with previous observations [31]. Large deviations from experiment (> 0.030 Å) are also observed at the UMP2 and RMP2 levels of theory for  and
and  . In these two cases, the spincontamination is small and the UMP2 and RMP2 geometries, although differing significantly from experiment, are quite similar.
. In these two cases, the spincontamination is small and the UMP2 and RMP2 geometries, although differing significantly from experiment, are quite similar.
As the data in Tables 6.4 and 6.5 indicate, all theoretical levels generally perform well in predicting bond angles at the radical center. MADs range from 0.3° to 0.9° while LDs range from -1.5° to +1.3°.
Overall, among the selected methods, the URCCSD(T)/cc-pVTZ procedure gives the best geometries for the radicals in Tables 6.2 - 6.5. The UB3LYP/6-31G(d) and UB3LYP/cc-pVTZ levels of theory also perform well, and are reasonably economical. The UMP2(fu)/6-31G(d) and RMP2/6-31G(d) approaches generally give acceptable geometries but are not reliable for radicals that display significant spin contamination. This may lead to occasional problems in the calculation of heats of formation for methods that use UMP2 geometries.

Theoretical Thermochemistry of Radicals |
169 |
4.HEATS OF FORMATION
The accurate prediction of the heats of formation of molecules has long been one of the main objectives of ab initio molecular orbital procedures [5, 32]. This is particularly important in radical chemistry, where it can be difficult to obtain accurate experimental results. A number of procedures have been used to obtain heats of formation at 0 K  from calculated total energies E [33]. We will illustrate them here using the ethyl radical
from calculated total energies E [33]. We will illustrate them here using the ethyl radical  as an example.
as an example.
In the atomization approach, the heat of formation of the radical is obtained by combining the calculated energy of the atomization reaction,
with the well-established heats of formation of the gaseous atoms to give

170 |
Chapter 6 |
In the formation approach, the heat of formation of the radical is obtained from the calculated energy of the formation reaction,
as
where the energy of the solid state carbon has been replaced by the difference 

Theoretical Thermochemistry of Radicals |
171 |
It has been found, for G2 calculations on organic molecules in particular, that the atomization method performs somewhat better than the formation method [33].
In the isodesmic approach, the heat of formation of the radical is obtained by combining the calculated energy of an appropriate isodesmic reaction involving the radical, e.g.
with accurate experimental heats of formation of the other species involved in the reaction,

172 |
Chapter 6 |
Because of cancellation of errors in reactions such as (4.5), reasonable results are often obtained, even at quite simple levels of theory. However, it has been found [21, 34] that larger errors may occur with unrestricted methods if there is a significant difference between the degrees of spin contamination for the two radicals in the reaction.
Theoretical Thermochemistry of Radicals |
173 |
Tables 6.6 - 6.8 present calculated [22] and experimental [3, 26, 35] heats of formation for a selection of small radicals, determined with the atomization approach. As these data show, all theoretical levels give good overall performance (MADs of 2.1 - 4.5 kJ/mol).  is the highest level of theory represented in these tables and indeed performs very well, with an MAD of 3.2 kJ/mol and an LD of +6.8 kJ/mol. The G2-RAD(QCISD) method gives the best statistical performance with an MAD of 2.1 kJ/mol and an LD of +5.6 kJ/mol. However, because of the modest number of comparisons, such differences are only of marginal significance. The G3-RAD approach also performs particularly well with an MAD of 2.5 kJ/mol and an LD of -5.5 kJ/mol. Overall, the G2RAD(QCISD) method tends to slightly overestimate the heats of formation of the selected radicals (MD of +0.8 kJ/mol) while its G3-RAD counterpart tends to underestimate them (MD of -2.0 kJ/mol). Both of these modified Gn methods offer improved performance over the standard G2 and G3 procedures. However, the G3(MP2)-RAD approach performs less well (MAD of 4.5 kJ/mol) than G3(MP2) (MAD of 3.6 kJ/mol).
is the highest level of theory represented in these tables and indeed performs very well, with an MAD of 3.2 kJ/mol and an LD of +6.8 kJ/mol. The G2-RAD(QCISD) method gives the best statistical performance with an MAD of 2.1 kJ/mol and an LD of +5.6 kJ/mol. However, because of the modest number of comparisons, such differences are only of marginal significance. The G3-RAD approach also performs particularly well with an MAD of 2.5 kJ/mol and an LD of -5.5 kJ/mol. Overall, the G2RAD(QCISD) method tends to slightly overestimate the heats of formation of the selected radicals (MD of +0.8 kJ/mol) while its G3-RAD counterpart tends to underestimate them (MD of -2.0 kJ/mol). Both of these modified Gn methods offer improved performance over the standard G2 and G3 procedures. However, the G3(MP2)-RAD approach performs less well (MAD of 4.5 kJ/mol) than G3(MP2) (MAD of 3.6 kJ/mol).
The CBS methods all give similar performance, with MADs of 3.2 - 3.6 kJ/mol. The CBS-Q and CBS-RAD variants tend to overestimate the selected radical heats of formation (MDs of +0.3 and +1.0 kJ/mol, respectively), while the CBS-QB3 procedure tends to slightly underestimate them (MD of -0.2 kJ/mol).
With the exception of one G2 case, all levels of theory predict the heats of formation of the selected radicals to within chemical accuracy (i.e.  ). The exceptional case is the ethynyl radical, which shows a deviation from experiment of 15.5 kJ/mol at the G2 level of theory. At the G2-RAD(QCISD) level this is reduced to 4.5 kJ/mol. The ethynyl radical exhibits significant spin contamination at the UMP2(fu)/6-31G(d) level and, as noted in the previous section, this leads to a poor geometry (Table 6.2). This is the major cause of the difference between the G2 and G2-RAD(QCISD) values. Similar lowerings of energies (and hence heats of formation) are observed upon going from G3 and G3(MP2) to G3-RAD and G3(MP2)-RAD, respectively. A similar situation is observed for the
). The exceptional case is the ethynyl radical, which shows a deviation from experiment of 15.5 kJ/mol at the G2 level of theory. At the G2-RAD(QCISD) level this is reduced to 4.5 kJ/mol. The ethynyl radical exhibits significant spin contamination at the UMP2(fu)/6-31G(d) level and, as noted in the previous section, this leads to a poor geometry (Table 6.2). This is the major cause of the difference between the G2 and G2-RAD(QCISD) values. Similar lowerings of energies (and hence heats of formation) are observed upon going from G3 and G3(MP2) to G3-RAD and G3(MP2)-RAD, respectively. A similar situation is observed for the  radical, for which the UMP2(fu)/6-31G(d) geometry is markedly inferior to those computed with the UB3LYP/6-31G(d) and UQCISD/6-31G(d) approaches.
radical, for which the UMP2(fu)/6-31G(d) geometry is markedly inferior to those computed with the UB3LYP/6-31G(d) and UQCISD/6-31G(d) approaches.
It was also noted in the previous section that the UMP2(fu)/6- 31G(d) level of theory performs badly in predicting the geometry of the  radical cation while the UB3LYP/6-31G(d) procedure performs quite well. This geometry difference makes a significant contribution to
radical cation while the UB3LYP/6-31G(d) procedure performs quite well. This geometry difference makes a significant contribution to
the difference between the G3 and G3-RAD heats of formation for