

2 |
Chapter 1 |
Nevertheless, in spite of all these efforts, it has proved exceedingly difficult to compute the necessary quantities (i.e., total electronic energies) to a target accuracy of better than 1 kJ/mol, which is the typical accuracy of experimental measurements of heats of formation. In this chapter, we examine the ab initio calculation of atomization energies (AEs) of gasphase molecules, from which the heats of gas-phase reactions between the same molecules can be easily obtained. Our purpose is not only to illustrate the inherent difficulties associated with the accurate calculation of AEs, but also to describe the considerable progress that has been achieved over the last few years and the perspectives for the near future.
2.HIERARCHIES OF AB INITIO THEORY
An important characteristic of ab initio computational methodology is the ability to approach the exact description – that is, the focal point [11] – of the molecular electronic structure in a systematic manner. In the standard approach, approximate wavefunctions are constructed as linear combinations of antisymmetrized products (determinants) of oneelectron functions, the molecular orbitals (MOs). The quality of the description then depends on the basis of atomic orbitals (AOs) in terms of which the MOs are expanded (the one-electron space), and on how linear combinations of determinants of these MOs are formed (the n- electron space). Within the oneand n-electron spaces, hierarchies exist of increasing flexibility and accuracy. To understand the requirements for accurate calculations of thermochemical data, we shall in this section consider the oneand n-electron hierarchies in some detail [12].
2.1.The Coupled-Cluster Hierarchy of n-Electron Models
At the lowest level of the n-electron hierarchy, we have the HartreeFock wavefunction, obtained by variationally optimizing a single determinant with respect to the shape of the occupied MOs. As we shall see, the quality of the single-determinant Hartree-Fock description is too low to provide sufficiently accurate thermochemical data. The inadequacy of the Hartree-Fock model arises from the fact that a single determinant gives an uncorrelated description of the electronic motion, in which each electron moves in the average (rather than instantaneous) field generated by the other electrons. This model is therefore incapable of describing the subtle changes that occur as electron pairs are broken or formed dur-

Highly Accurate Ab Initio Computation of Thermochemical Data |
3 |
ing chemical processes. For an accurate description of thermochemical data, we must therefore go beyond the Hartree-Fock level of theory.
A standard method of improving on the Hartree-Fock description is the coupled-cluster approach [12, 13]. In this approach, the wavefunction  is written as an exponential of a cluster operator
is written as an exponential of a cluster operator  working on the Hartree-Fock state
working on the Hartree-Fock state  generating a linear combination of all possible
generating a linear combination of all possible
determinants that may be constructed in a given one-electron basis,
The cluster operator  creates excitations out of the Hartree-Fock determinant and may be written as
creates excitations out of the Hartree-Fock determinant and may be written as
where  creates single excitations,
creates single excitations,  creates double excitations, and so on. There is ample theoretical and numerical evidence that the contributions from
creates double excitations, and so on. There is ample theoretical and numerical evidence that the contributions from  rapidly decrease after
rapidly decrease after  (double excitations), and the coupled-cluster operator is therefore usually truncated either after
(double excitations), and the coupled-cluster operator is therefore usually truncated either after  leading to the coupled-cluster singles-and-doubles (CCSD) model, or after
leading to the coupled-cluster singles-and-doubles (CCSD) model, or after  leading to the CCSDT method, including all singles, doubles, and triples. The advantage of the exponential parameterization is that the wavefunction becomes multiplicatively separable, thereby providing a uniform (size-extensive) description of systems of different size. Other hierarchies of n-electron models exist, but the coupled-cluster hierarchy has proved to be the most successful for highly accurate calculations of molecular electronic structure. In particular, the configurationinteraction (CI) method suffers from a lack of size-extensivity and the related slow convergence with respect to the level of excitation; the perturbative Møller-Plesset method is not sufficiently accurate to low orders and converges slowly if at all [14].
leading to the CCSDT method, including all singles, doubles, and triples. The advantage of the exponential parameterization is that the wavefunction becomes multiplicatively separable, thereby providing a uniform (size-extensive) description of systems of different size. Other hierarchies of n-electron models exist, but the coupled-cluster hierarchy has proved to be the most successful for highly accurate calculations of molecular electronic structure. In particular, the configurationinteraction (CI) method suffers from a lack of size-extensivity and the related slow convergence with respect to the level of excitation; the perturbative Møller-Plesset method is not sufficiently accurate to low orders and converges slowly if at all [14].
To understand the structure of the coupled-cluster wavefunction, let us Taylor expand the exponential in Eq. (2.1). Sorting the resulting expansion according to the level of excitation, we obtain
At each excitation level beyond the single-excitation level, a number of terms contribute. For example, double excitations are generated both by means of the double-excitation operator  (connected excitations)
(connected excitations)
4 |
Chapter 1 |
and by means of two independent single-excitation operators  (disconnected excitations). At the CCSD level, we ignore all triple and higher connected excitations. Nevertheless, because of the presence of disconnected contributions in Eq. (2.3), the resulting CCSD wavefunction contains contributions from all levels of excitation – that is, it contains contributions from all determinants that can be constructed in a given AO basis. Since the higher excitations are dominated by disconnected contributions, this separation into connected and disconnected contributions ensures both a rapid convergence of the coupled-cluster hierarchy and size-extensivity of its energy.
(disconnected excitations). At the CCSD level, we ignore all triple and higher connected excitations. Nevertheless, because of the presence of disconnected contributions in Eq. (2.3), the resulting CCSD wavefunction contains contributions from all levels of excitation – that is, it contains contributions from all determinants that can be constructed in a given AO basis. Since the higher excitations are dominated by disconnected contributions, this separation into connected and disconnected contributions ensures both a rapid convergence of the coupled-cluster hierarchy and size-extensivity of its energy.
2.2.The Correlation-Consistent Hierarchy of One-Electron Basis Sets
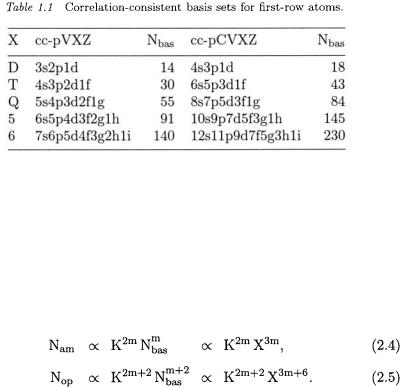
The quality of quantum-chemical calculations depends not only on the chosen n-electron model but also critically on the flexibility of the one-electron basis set in terms of which the MOs are expanded. Obviously, it is possible to choose basis sets in many different ways. For highly accurate, systematic studies of molecular systems, it becomes important to have a well-defined procedure for generating a sequence of basis sets of increasing flexibility. A popular hierarchy of basis functions are the correlation-consistent basis sets of Dunning and coworkers [1517]. We shall use two varieties of these sets: the cc-pVXZ (correlationconsistent polarized-valence X-tuple-zeta) and cc-pCVXZ (correlationconsistent polarized core-valence X-tuple-zeta) basis sets; see Table 1.1.
As can be seen from the table, the number of AOs increases rapidly with the cardinal number X. Thus, with each increment in the cardinal number, a new shell of valence AOs is added to the cc-pVXZ set; since the number of AOs added in each step is proportional to  the total number
the total number  of AOs in a correlation-consistent basis set is proportional to
of AOs in a correlation-consistent basis set is proportional to  The core-valence sets cc-pCVXZ contain additional AOs for the correlation of the core electrons. As we shall see later, the hierarchy of correlation-consistent basis sets provides a very systematic description of molecular electronic systems, enabling us to develop a useful extrapolation technique for molecular energies.
The core-valence sets cc-pCVXZ contain additional AOs for the correlation of the core electrons. As we shall see later, the hierarchy of correlation-consistent basis sets provides a very systematic description of molecular electronic systems, enabling us to develop a useful extrapolation technique for molecular energies.
Highly Accurate Ab Initio Computation of Thermochemical Data |
5 |
2.3.Computational Cost
The computational complexity of the coupled-cluster method truncated after a given excitation level m – for example,  for CCSD
for CCSD
– may be discussed in terms of the number of amplitudes  in the coupled-cluster operator and the number of operations
in the coupled-cluster operator and the number of operations  required for optimization of the wavefunction. Considering K atoms, each with
required for optimization of the wavefunction. Considering K atoms, each with
 basis functions, we have the following scaling relations:
basis functions, we have the following scaling relations:
From these expressions, it is evident that the steep increase of the computational cost with X (the basis-set hierarchy) and m (the coupled-cluster hierarchy) severely restricts the levels of theory that can be routinely used for large systems or even explored for small systems. Nevertheless, we shall see that, with current computers, it is possible to arrange the calculations in such a manner that chemical accuracy (of the order of  ) can be achieved – at least for molecules containing not more than ten first-row atoms.
) can be achieved – at least for molecules containing not more than ten first-row atoms.
3.CONVERGENCE OF THE COUPLED-CLUSTER HIERARCHY
3.1. Model Calculations on  and HF
and HF
For small basis sets and molecules, it is possible to calculate the full set of energies in the coupled-cluster hierarchy, from the Hartree-Fock to the full configuration-interaction (FCI) energy [18]. Although such

6 |
Chapter 1 |
calculations employ basis sets that are too small to give quantitative estimates of AEs, they still provide useful information about the convergence of the coupled-cluster hierarchy – in particular, about the importance of the quadruple and higher excitations. We here present two such series of calculations.
In Table 1.2, we have listed the valence cc-pVDZ electronic energies and AEs of  and HF at different levels of coupled-cluster theory. The energies are given as deviations from the FCI values. Comparing the different levels of theory, we note that the error is reduced by one order of magnitude at each level. In particular, at the CCSDT level, there is a residual error of the order of a few kJ/mol in the calculated energies and AEs, suggesting that the CCSDTQ model is usually needed to reproduce experimental measurements to within the quoted errors bars (often less than 1 kJ/mol).
and HF at different levels of coupled-cluster theory. The energies are given as deviations from the FCI values. Comparing the different levels of theory, we note that the error is reduced by one order of magnitude at each level. In particular, at the CCSDT level, there is a residual error of the order of a few kJ/mol in the calculated energies and AEs, suggesting that the CCSDTQ model is usually needed to reproduce experimental measurements to within the quoted errors bars (often less than 1 kJ/mol).
Let us make two more observations about the convergence of the coupled-cluster hierarchy. First, it converges faster for HF than for  reflecting the more complicated electronic structure of the multiple bond in
reflecting the more complicated electronic structure of the multiple bond in  Second, the cancellation of errors in the calculated AEs becomes less pronounced as we move up in the coupled-cluster hierarchy. The cancellation diminishes since the coupled-cluster expansions of the atoms
Second, the cancellation of errors in the calculated AEs becomes less pronounced as we move up in the coupled-cluster hierarchy. The cancellation diminishes since the coupled-cluster expansions of the atoms