

BDEs of Transition Metal Compounds and Main Group Complexes |
221 |
The calculations indicate that the  BDEs of the latter complexes are 10 - 12 kcal/mol lower than those of the former ones. The second carbonyl ligand that is in the trans position with respect to the boryl group in
BDEs of the latter complexes are 10 - 12 kcal/mol lower than those of the former ones. The second carbonyl ligand that is in the trans position with respect to the boryl group in  has a rather low BDE. This result may be expected since both classes of complexes coexist and can be interconverted by changing the CO pressure [83]. Table 7.15 also lists the calculated
has a rather low BDE. This result may be expected since both classes of complexes coexist and can be interconverted by changing the CO pressure [83]. Table 7.15 also lists the calculated  BDEs. These bond energies are significantly higher that those of the osmium compounds, although boron generally forms stronger bonds with transition metals than gallium. The high bond energy may partly be explained by the effect of the ammonia ligand that has been already shown to enhance the bond strength of the M–E bond, when M is a group-13 element.
BDEs. These bond energies are significantly higher that those of the osmium compounds, although boron generally forms stronger bonds with transition metals than gallium. The high bond energy may partly be explained by the effect of the ammonia ligand that has been already shown to enhance the bond strength of the M–E bond, when M is a group-13 element.
12.TRANSITION METAL METHYL AND PHENYL COMPOUNDS
Another class of M–ligand bonds for which BDEs have been calculated are methyl and phenyl bonds of the group-11 metals Cu, Ag, Au and the group-12 metals Zn, Cd and Hg [28]. Table 7.16 compiles the predicted BDEs calculated at the MP2/II and CCSD(T)/II levels of theory as well as the relevant experimental data.
The most interesting conclusion from the results shown in Table 7.16 is that, except for 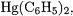 the MP2/II BDEs are in excellent agreement with the experimental data. Although the good agreement between the MP2/II calculations and experiment is due to a fortuitous error cancellation, the question remains why the theoretical value for
the MP2/II BDEs are in excellent agreement with the experimental data. Although the good agreement between the MP2/II calculations and experiment is due to a fortuitous error cancellation, the question remains why the theoretical value for
 is too high when all the other values are correct. The calculations predict that the metal–phenyl bonds have higher BDEs than the metal–methyl bonds, while the only experimental value available for a phenyl compound of a group-12 element predicts the opposite. Since
is too high when all the other values are correct. The calculations predict that the metal–phenyl bonds have higher BDEs than the metal–methyl bonds, while the only experimental value available for a phenyl compound of a group-12 element predicts the opposite. Since
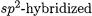 bonds of carbon are normally stronger than their
bonds of carbon are normally stronger than their 
 counterparts, we tend to believe that the theoretical value
counterparts, we tend to believe that the theoretical value
is correct. Unfortunately, CCSD(T)/II calculations of  could not be carried out because the molecule was too big. The BDEs predicted at the CCSD(T)/II level of theory are slightly lower than both their MP2/II and experimental counterparts but the trend is the same.
could not be carried out because the molecule was too big. The BDEs predicted at the CCSD(T)/II level of theory are slightly lower than both their MP2/II and experimental counterparts but the trend is the same.

222 |
Chapter 7 |
13.TRANSITION METAL NITRIDO AND PHOSPHIDO COMPLEXES
The nitrido and phosphido complexes of TMs have been the subjects of intensive experimental studies in the recent years. Of particular interest has been the issue of Lewis basicity of the nitrogen and phosphorus atoms in the  and
and 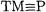 groups. Table 7.17 lists the BDEs calculated at the MP2/II, B3LYP/II and CCSD(T)/II levels of theory for
groups. Table 7.17 lists the BDEs calculated at the MP2/II, B3LYP/II and CCSD(T)/II levels of theory for  and
and  where X is a group-13 Lewis acid or a chalcogen atom [86, 87].
where X is a group-13 Lewis acid or a chalcogen atom [86, 87].
These results demonstrate that it is difficult to make a general statement about the accuracy of the MP2 or B3LYP approaches vis-a-vis that of CCSD(T). For example, the BDEs of the  donor-acceptor bonds (E = B, Al, Ga) are very similar at the MP2/II and B3LYP/II levels of theory. They also agree with the CCSD(T)/II value for the N–
donor-acceptor bonds (E = B, Al, Ga) are very similar at the MP2/II and B3LYP/II levels of theory. They also agree with the CCSD(T)/II value for the N–
 bond. However, for the
bond. However, for the  and
and  bonds, the B3LYP/II
bonds, the B3LYP/II

BDEs of Transition Metal Compounds and Main Group Complexes |
223 |
level of theory produces significantly lower BDEs than the MP2/II one.
Since the CCSD (T) /II values for the |
and |
BDEs coincide |
with the MP2/II estimates, we believe that for the |
bonds the |
|
latter are more reliable than their B3LYP/II counterparts. On the other hand, the two methods yield nearly identical BDEs for the N–X bonds, where X is a chalcogen.
The results for the P–S BDEs in the phosphido complexes are even more confusing. The B3LYP/II predictions are significantly larger than the MP2/II ones, while the CCSD(T)/II level of theory produces intermediate values of BDEs. Moreover, it has been found that the MP2/II BDEs for the molybdenum complexes are lower than those for the tungsten complexes, while the predictions of the B3LYP/II and CCSD(T)/II levels of theory are that the BDEs of the MoP–S bonds are significantly higher than those of the WP–S bonds. The MP2/II BDEs of the Mo–

224 |
Chapter 7 |
 and
and  donor-acceptor bonds appear to be more reliable as they agree quite well with the CCSD(T)/II estimates.
donor-acceptor bonds appear to be more reliable as they agree quite well with the CCSD(T)/II estimates.
14.MAIN GROUP COMPLEXES OF GROUP-13 LEWIS ACIDS 
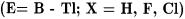
The nature of the donor-acceptor interactions in main group complexes has also been studied theoretically [88-91]. The calculated BDEs of main-group complexes predicted at the MP2/II level of theory are in very good agreement with experimental results [88]. This is an important difference from the performance of the MP2/II level of theory for TM complexes, where the computed BDEs are always too high. Table 7.18 lists the calculated BDEs for complexes of group-13 Lewis acids  with various Lewis bases.
with various Lewis bases.
A comparison of the calculated and experimental data indicates that the MP2 values obtained with the basis sets II do not differ significantly from those afforded by the larger basis set TZP. The theoretical values agree very well with the experimental data. The only larger deviation from experimental data has been found for  (Table 7.18). However, a critical examination of the experimental value of 30.5 kcal/mol led us to suggest that it is probably too low [88].
(Table 7.18). However, a critical examination of the experimental value of 30.5 kcal/mol led us to suggest that it is probably too low [88].
On the basis of the calculated BDEs, it possible to estimate the strength of the Lewis acidity of the  compounds. The heavier group13 trifluorides and trichlorides of Al, Ga, and In exhibit similar Lewis acidities and they are generally stronger Lewis acids than
compounds. The heavier group13 trifluorides and trichlorides of Al, Ga, and In exhibit similar Lewis acidities and they are generally stronger Lewis acids than 
and  However, the bond strengths of donor-acceptor complexes depend also on the nature of the Lewis base. For example, the MP2/TZP BDE of
However, the bond strengths of donor-acceptor complexes depend also on the nature of the Lewis base. For example, the MP2/TZP BDE of  is only 2.3 kcal/mol, that is markedly less than the
is only 2.3 kcal/mol, that is markedly less than the  BDE of 15.1 kcal/mol, while the
BDE of 15.1 kcal/mol, while the  BDE of 59.7 kcal/mol is slightly higher than that of
BDE of 59.7 kcal/mol is slightly higher than that of  namely 58.0
namely 58.0
kcal/mol. However, the Lewis acid strengths of boron compounds are generally lower than those of their heavier analogues. The calculations indicate that the Lewis acidity of the former species exhibits the trend

BDEs of Transition Metal Compounds and Main Group Complexes |
225 |