

BDEs of Transition Metal Compounds and Main Group Complexes |
207 |
4.IRON CARBONYL COMPLEXES 
A large number of iron carbonyl complexes 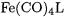 with different ligands L in the axial or equatorial positions has been investigated [58]. Table 7.5 lists the theoretically predicted relative energies of the isomers and the Fe–L BDEs at the B3LYP/II and CCSD(T)/II levels of theory. Experimental values of these bond energies are not known. The
with different ligands L in the axial or equatorial positions has been investigated [58]. Table 7.5 lists the theoretically predicted relative energies of the isomers and the Fe–L BDEs at the B3LYP/II and CCSD(T)/II levels of theory. Experimental values of these bond energies are not known. The  complexes, where L is a group-13 diyl ligand, are presented separately.
complexes, where L is a group-13 diyl ligand, are presented separately.
The data compiled in Table 7.5 show that the relative energies of the axial and equatorial isomers yielded by the B3LYP/II level of theory are very similar to their CCSD(T) counterparts. The B3LYP/II BDEs are always larger than the CCSD(T)/II results, but the trends predicted by the two methods for different ligands are the same. It is worth noting

208 |
Chapter 7 |
that the B3LYP/II and CCSD(T)/II levels of theory agree on ethylene in  being more strongly bonded than acetylene.
being more strongly bonded than acetylene.

BDEs of Transition Metal Compounds and Main Group Complexes |
209 |
5. GROUP-10 CARBONYL COMPLEXES 
Table 7.6 lists the theoretical BDEs of the M–L bonds in the group-  and
and  complexes calculated at the MP2/II and CCSD(T)/II levels of theory [49, 50]. The only experimental value known for those compounds is an estimate of ca. 10 kcal/mol obtained for the
complexes calculated at the MP2/II and CCSD(T)/II levels of theory [49, 50]. The only experimental value known for those compounds is an estimate of ca. 10 kcal/mol obtained for the  bond energy at 298 K [59]. This estimate is based on kinetic measurements of nitrogen extrusion from the complex. Thermal corrections to the CCSD(T)/II value of
bond energy at 298 K [59]. This estimate is based on kinetic measurements of nitrogen extrusion from the complex. Thermal corrections to the CCSD(T)/II value of  kcal/mol yield a theoretical prediction of 6.7 kcal/mol, which is in a reasonable agreement with experiment [49]. The MP2/II BDEs listed in
kcal/mol yield a theoretical prediction of 6.7 kcal/mol, which is in a reasonable agreement with experiment [49]. The MP2/II BDEs listed in

210 |
Chapter 7 |
Table 7.6 are significantly larger than their CCSD(T)/II counterparts.  is much more strongly bonded at the MP2/II level of theory (
is much more strongly bonded at the MP2/II level of theory ( kcal/mol) than at the CCSD(T)/II level. The stronger metal–ligand bonds at the MP2/II level of theory lead to energy minima for the
kcal/mol) than at the CCSD(T)/II level. The stronger metal–ligand bonds at the MP2/II level of theory lead to energy minima for the  complexes that are predicted by the CCSD(T)/II calculations to be unstable with respect to the M–L dissociation. For example, both
complexes that are predicted by the CCSD(T)/II calculations to be unstable with respect to the M–L dissociation. For example, both  and
and  are minima on the MP2/II potential energy hypersurfaces, while the calculations at the CCSD(T)/II level of theory produce negative dissociation energies [42]. The M–L BDEs of the group-10 carbonyl complexes
are minima on the MP2/II potential energy hypersurfaces, while the calculations at the CCSD(T)/II level of theory produce negative dissociation energies [42]. The M–L BDEs of the group-10 carbonyl complexes  are clearly lower than those of their group-6 analogues
are clearly lower than those of their group-6 analogues  They are also lower than the Fe–L BDEs of
They are also lower than the Fe–L BDEs of 
6.GROUP-6 CARBONYL COMPLEXES WITH PHOSPHANE LIGANDS 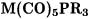
The structure and bonding of group-6 TM carbonyl complexes  with phosphane ligands
with phosphane ligands  and
and  have been the subjects of another theoretical study [60]. Table 7.7 lists the
have been the subjects of another theoretical study [60]. Table 7.7 lists the  BDEs calculated at the BP86 level of theory in conjunction with our standard basis set II and the larger TZ(2)P Slater basis set, which
BDEs calculated at the BP86 level of theory in conjunction with our standard basis set II and the larger TZ(2)P Slater basis set, which
has one set of f-type |
polarization functions on the transition metals and |
two sets of polarization functions on the other atoms. |
|
Except for the |
complexes, the BP86/II and BP86/TZ(2)P |
BDEs values are very similar. The BP86/II BDE estimates for these complexes are 4 - 6 kcal/mol higher than their BP86/TZ(2)P counter-
parts. Both levels of theory predict the trend in the |
BDEs for |
the different phosphane ligands being |
|
7.NOBLE GAS COMPLEXES 
A special type of TM ligands are the noble gas atoms argon, krypton, and xenon [61]. Although they are weak Lewis bases, TM complexes  with
with 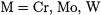 and
and  and Xe have been experimentally investigated in the gas phase as well as in the liquid phase and in supercritical
and Xe have been experimentally investigated in the gas phase as well as in the liquid phase and in supercritical  The M–Ng BDEs were estimated with
The M–Ng BDEs were estimated with