

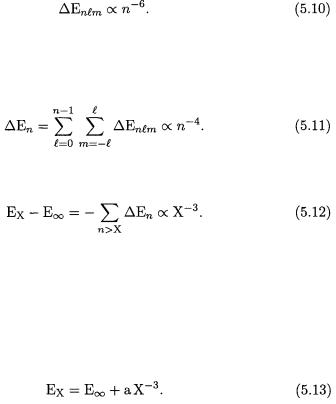
Highly Accurate Ab Initio Computation of Thermochemical Data |
15 |
The use of the linear  term for many-electron systems is more involved but has been successfully incorporated in the framework of coupled-cluster theory by Kutzelnigg and coworkers [43-46]. In their R12 theory, the difficult many-electron integrals that arise from the inclusion of the interelectronic distance in the wavefunction are avoided by the resolution-of-identity approximation, yielding a highly efficient scheme for the accurate calculation of atomic and molecular electronic energies. For example, comparing with standard coupled-cluster calculations, the evaluation of the R12 two-electron integrals requires only about four times more computation time; the remaining part of the calculation requires essentially no additional computational effort.
term for many-electron systems is more involved but has been successfully incorporated in the framework of coupled-cluster theory by Kutzelnigg and coworkers [43-46]. In their R12 theory, the difficult many-electron integrals that arise from the inclusion of the interelectronic distance in the wavefunction are avoided by the resolution-of-identity approximation, yielding a highly efficient scheme for the accurate calculation of atomic and molecular electronic energies. For example, comparing with standard coupled-cluster calculations, the evaluation of the R12 two-electron integrals requires only about four times more computation time; the remaining part of the calculation requires essentially no additional computational effort.
5.2.Extrapolations from Principal Expansions
Although the convergence of the FCI principal expansion is slow, it is systematic [12]. In fact, for a sufficiently large basis, it has been found that each STO in the FCI principal expansion of He contributes an amount of energy that, to a good approximation, is given by the expression [47, 48]
Note that the energy contribution depends only on the principal quantum number n. Therefore, each of the  orbitals that constitute the shell n contributes the same amount of energy, justifying the use of the principal expansion. Summing the energy contributions from all orbitals, we obtain
orbitals that constitute the shell n contributes the same amount of energy, justifying the use of the principal expansion. Summing the energy contributions from all orbitals, we obtain
By summing the contributions from all neglected shells, it is now easy to estimate the error that arises when the principal expansion is truncated after 
This empirical result is consistent with the theoretical analysis of the partial-wave expansion (where the truncation of the FCI expansion is based on the angular-momentum quantum number 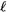 rather than on the principal quantum number n), for which it has been proved that the truncation error is proportional to
rather than on the principal quantum number n), for which it has been proved that the truncation error is proportional to  when all STOs up to
when all STOs up to  are included in the FCI wavefunction [49, 50].
are included in the FCI wavefunction [49, 50].
Guided by Eq. (5.12), we assume that the calculated He energy is well represented by the expression