

1.4.5. Квантово-химические расчеты
Используются две группы методов: неэмпирические – ab initio и полуэмпирические. Наиболее точные результаты дает ab initio. Оба используют SCF MO self-consistent-field molecular orbitals, т. е. метод самосогласованного поля и приближения молекулярных орбиталей.
В неэмпирических методах вычисляют большое число молекулярных интегралов. С увеличением размера молекулы базис расчета возрастает приблизительно пропорционально n4. Величина n – общее число базисных атомных орбиталей (АО). При учете конфигурационных взаимодействий, электронной корреляции, поправок на релятивистские эффекты и неадиабатичность существенно увеличивается время расчета. В простейшем случае неэмпирические расчеты строятся с использованием минимального базиса АО. Каждая из АО в разложении представлена одной орбиталью слейтеровского типа (STO). Минимальный базис, содержащий N гауссовских функций (GTO), примененный для аппроксимации одной слейтеровской, равняется трем – STO-3G.
Из-за больших затрат времени на расчеты обычно используют полуэмпирические методы. Разница в том, что в полуэмпирических методах рассматривают только взаимодействия валентных электронов, при этом некоторая часть этих интегралов параметризуется и/или задается константами (именно это и является причиной потери точности), тогда как в ab initio все эти взаимодействия просчитываются (отсюда и большие затраты времени). Приближения, используемые в полуэмпирических методах:
- CNDO complete neglect of differential overlap (Pople, Santry, Segal/1965) рассматривает только валентные электроны. Метод аппроксимирован на атомных потенциалах и сродстве к электрону, причем некоторые из них брались как полуэмпирические величины. Первый метод назывался CNDO/1; в настоящее время используется CNDO/2, в нем улучшена параметризация.
- MINDO/1 (Barid, Dewar/1969) – первая версия модифицированного INDO. Параметры выбирались на основе экспериментальных молекулярных теплот образования, но метод плохо воспроизводил геометрию молекулы. Поэтому в 1970 г. был улучшен в методе MINDO/2 (дает хорошие результаты по длинам связей и теплотам образования, но плохие по длинам связей с водородом). Следующие версии были MINDO/2’ и MINDO/3. Наиболее удачен последний, его ошибки составили 11 ккал/моль по теплоте образования, 0,02 Å по длинам связей, 5 по углам, 0,4D по дипольному моменту и 0,8 эВ по величинам потенциалов ионизации.
- MNDO (модифицированный MNDO) разработан в 1977 г. Его ошибки по величинам: по теплоте образования 9 ккал/моль, 0,025 Å по длинам связей, 3 по углам, 0,36 D по дипольному моменту и 0,5 эВ по потенциалу ионизации.
- AM1 (Austin Model 1) разработан на основе MNDO в 1985 г. В нем была улучшена способность воспроизведения водородных связей, по сравнению с предшествующей версией.
- PM/3 (MNDO parametric method 3) (первый и второй были MNDO и AM1) существенно снизил ошибки MNDO и AM1, при этом точность расчетов теплот образования, геометрии, дипольного момента не теряется.
Сравнение методов MNDO, AM1, РМ3 показывает то, что абсолютная ошибка по Hf298 составляет 22,5 %, 13,8 % и 8,2 % соответственно.
В тонком органическом синтезе для оценки направления атаки реагента на один из альтернативных реакционных центров используют величины зарядов на них (зарядовый контроль реакции), величины и знаки коэффициентов атомной орбитали (АО) высшей занятой молекулярной орбитали или низшей свободной орбитали – АОВЗМО или АОНСМО (орбитальный контроль реакции). Для сравнения реакционной способности в ряду соединений важно знать и величины энергии граничных орбиталей ЕВЗМО и ЕНСМО. Эти данные могут быть получены с помощью квантово-химических расчетов. В реакциях электрофильного и нуклеофильного замещения в ряду ароматических и гетероароматических соединений наблюдается зарядовый контроль. В качестве примера на схеме приведены величины зарядов на атомах двух молекул (расчет проведен в приближении АМ1).
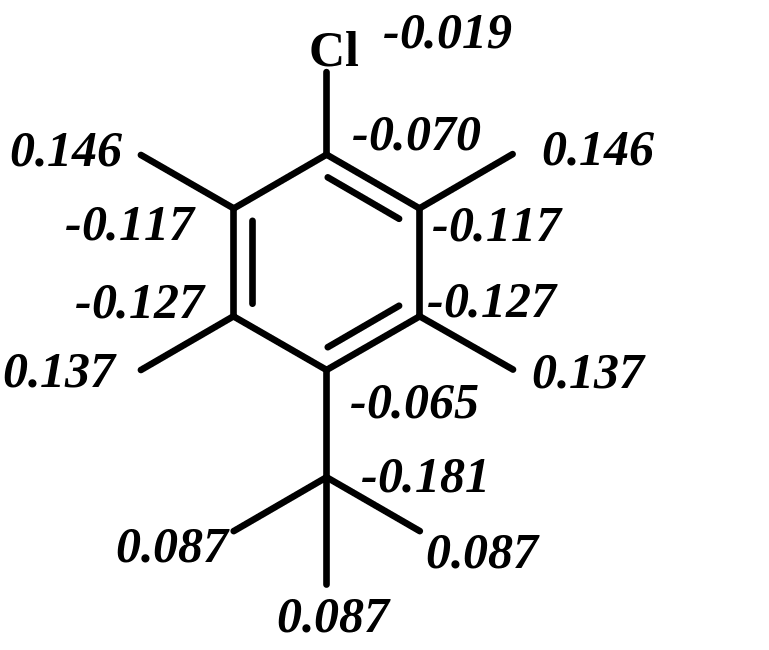
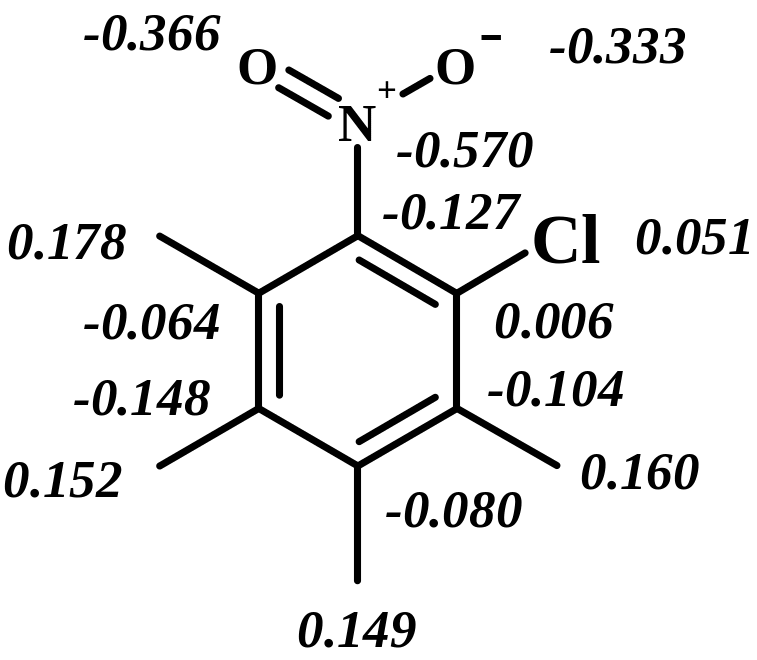
Дипольные моменты: 1,615D 5,351D
ЕВЗМО = - 9,296 эВ, ЕНСМО = 0,134 эВ ЕВЗМО = - 10,314 эВ, ЕНСМО = -1,263 эВ
Экспериментально найденные величины дипольных моментов данных молекул в газовой фазе составляют соответственно 1,74D и 4,35D, что близко к расчетным данным и подтверждает корректность полученных величин. Для сравнения дипольные моменты молекул, найденные расчетом по методу CNDO/2, составляют 3,151D и 6,72D. Сравнение расчетных величин дипольных моментов, полученных с помощью методов АМ1 и CNDO/2, с экспериментальными позволяет отдать предпочтение методу АМ1. Из величин зарядов на атомах углерода п-хлортолуола видно, что в реакциях электрофильного замещения атака электрофильного агента предпочтительнее в положение 2, хотя не исключается атака и в положение 3. Подробно поведение этого соединения в реакции нитрования будет рассмотрено в разделе 2.1.1. В случае о-нитрохлорбезола атака электрофильного агента предпочтительнее в положение четыре ароматического цикла. Действительно, в случае нитрования о-нитрохлорбезола в основном образуется 2,4-динитрохлорбензол. Если рассмотреть поведение этих соединений в реакциях нуклеофильного замещения, то из данных расчета видно, что при взаимодействии с нуклеофилами замещение атома хлора в о-нитрохлорбезоле возможно, а в п-хлортолуоле проблематично.
Таким образом, в сложных случаях с помощью квантово-химических расчетов можно прогнозировать поведение молекулы в различных реакциях. Однако корректность расчетов должна быть подтверждена соответствием полученных результатов с физическими параметрами. Например, с геометрией молекулы, найденной с помощью рентгеноструктурного анализа (РСА), а в приведенных примерах с величинами дипольных моментов молекул. В противном случае из результатов квантово-химических расчетов можно сделать неправильные выводы.
Квантово-химические расчеты используют также для описания механизма реакции. При этом они являются единственным некосвенным источником информации о структуре и энергетике переходных состояний, которые в принципе невозможно наблюдать экспериментально. Они также незаменимы при рассмотрении высоколабильных интермедиатов, поскольку экспериментальные методы не позволяют их охарактеризовать. В таких случаях ведут расчет поверхности потенциальной энергии (ППЭ).