

Контрольные вопросы.
Граница раздела фаз, ее особенности. Поверхностная энергия.
Какие явления называют сорбцией, адсорбцией, абсорбцией?
В чем заключается разница между адсорбцией и абсорбцией?
В чем заключается разница между физической адсорбцией и хемосорцией ?
От каких факторов зависит адсорбция?
Какие уравнения описывают адсорбцию?
Основные положения теории Ленгмюра, уравнение изотермы адсорбции и приведение его к линейному виду, экспериментальное определение констант уравнения изотермы.
Уравнение изотермы адсорбции Фрейндлиха, приведение его к линейному виду, экспериментальное определение констант в уравнении Фрейндлиха.
Теория полимолекулярной теории Поляни.
Уравнение изотермы адсорбции БЭТ и его практическое применение.
4.1 Анализ твердой поверхности на содержание кислотно-основных центров
Структура поверхности
Всякая реальная поверхность твердого тела, являясь, сама по себе одним из основных дефектов трехмерной структуры кристаллической решетки, неоднородна в геометрическом, химическом и электронном смысле. Обрыв периодичности решетки приводит к изменению координационной сферы поверхностных атомов и регибридизации связей, в результате чего изменяются эффективные заряды и порядок расположения атомов на поверхности, межатомные углы и расстояния. Кристаллографическая структура поверхности в принципе отлична от структуры в объеме.
Источники неоднородности в поверхности многообразны и многочисленны – это изломы, ступени, дефекты, дислокации, выход различных кристаллографических граней и элементов структурных ячеек, продукты адсорбции в результате предварительной обработки материала и его контакта с окружающей средой.
Нарушение хода периодического потенциала на поверхности, вызванное обрывом периодичности решетки, присутствием разнообразных макро- и микродефектов, а также различных поверхностных химических соединений приводит к появлению широкого спектра поверхностных состояний – центров адсорбции, С точки зрения современных представлений поверхность твердого тела – это обусловленный набор орбиталей. Их распределение по энергиям, индивидуальное для каждой поверхности в конкретных условиях, определяется составом энергетических уровней – вакантные, заселенные, одно- и многоэлектронные. Характер энергетической неоднородности поверхности определяет ее химические свойства, конкретизируя при этом спектр активных поверхностных центров, в качестве которых могут выступать как поверхностные атомы решетки с более или менее связанным электроном, так и поверхностные функционалы, образовавшиеся в результате взаимодействия с молекулами окружающей среды.
Состав и реакционная способность поверхности твердого вещества зависит от формы и размера частиц образца, пористости, структурной и кристаллографической модификации, природы и содержания примесей в объеме и на поверхности, температуры активации, способа производства, условий хранения, степени гидратации и т.д. Однако во всех случаях энергетические свойства поверхности следует рассматривать в ее взаимодействии со средой, которая формирует новую поверхность, изменяя состав и структуру поверхностных центров. Влияние неоднородности на активность поверхности – очень важный с технологической точки зрения вопрос.
Наиболее подробно исследована структура поверхности твердых оксидов, поскольку они характеризуются простым и однородным спектром поверхностных состояний. В общем случае на поверхности твердого оксида можно выделить следующие типы центров, с участием которых протекают процессы адсорбции:
электроноакцепторные орбитали катионов металла;
электронодонорные орбитали ионов кислорода;
гидроксильно-гидратный покров, образующийся при различных формах адсорбции молекул воды и ее фрагментов.
Теории кислот и оснований
Теории кислот и оснований – совокупность фундаментальных физико-химических представлений, описывающих природу и свойства кислот и оснований.
Теория электролитической диссоциации Аррениуса-Оствальда (1887). Кислоты – это вещества, образующие в водном растворе гидратированные катионы водорода Н+ (ионы гидроксония Н3О+) и анионы кислотного остатка; основания в водном растворе диссоциируют с образованием катионов металла и гидроксид-анионов ОН−. В общем случае, кислотно-основное взаимодействие сводится к нейтрализации с образованием соли и воды. Эта теория вполне удовлетворительно описывала свойства водных растворов кислот и оснований, гидролиз солей. На основе представлений о степени и константе диссоциации было закреплено деление электролитов на сильные и слабые, введено понятие водородного показателя. Вместе с тем, её весьма затруднительно использовать для неводных сред. Теория не позволяет объяснить наличие основных свойств аммиака, фосфина и других соединений, не содержащих гидроксогрупп.
Протолитическая (протонная) теория кислот и оснований Брёнстеда-Лоури (1923). В ней понятие о кислотах и основаниях было объединено в единое целое, проявляющееся в кислотно-основном взаимодействии: А ↔ В + Н+ (где А – кислота, В – основание). Согласно этой теории кислотами являются молекулы или ионы, способные быть в данной реакции донорами протонов, а основаниями являются молекулы или ионы, присоединяющие протоны (акцепторы). Кислоты и основания получили общее название протолитов.
Сущностью кислотно-основного взаимодействия является передача протона от кислоты к основанию. При этом кислота, передав протон основанию, сама становится основанием, так как может снова присоединять протон, а основание, образуя протонированную частицу, становится кислотой. Таким образом, в любом кислотно-основном взаимодействии участвуют две пары кислот и оснований, названные Бренстедом сопряженными: А1 + В2 ↔ А2 + В1.
Одно и то же вещество в зависимости от условий взаимодействия может быть как кислотой, так и основанием (амфотерность или амфолитность). Например, вода при взаимодействии с сильными кислотами является основанием: H2O + H+ ↔ H3О+, а реагируя с аммиаком, становится кислотой: NH3 + H2O ↔ NH4+ + OH−.
Электронная теория Льюиса (1923). На основе электронных представлений было ещё более расширено понятие кислоты и основания. Кислота Льюиса – молекула или ион, имеющие вакантные электронные орбитали, вследствие чего они способны принимать электронные пары. Это, например, ионы водорода – протоны, ионы металлов (Ag+, Fe3+), оксиды некоторых неметаллов (например, SO3, SiO2), ряд солей (AlCl3), а также такие вещества как BF3, Al2O3. Кислоты Льюиса, не содержащие ионов водорода, называются апротонными. Протонные кислоты рассматриваются как частный случай класса кислот. Основание Льюиса – это молекула или ион, способные быть донором электронных пар: все анионы, аммиак и амины, вода, спирты, галогены.
В середине 30-х годов Вернадский и Полинг показали, что поверхность твердого тела обладает свойствами кислот и оснований. Под твердой кислотой (основанием) подразумевается твердое тело, на котором происходит изменение цвета основного (кислотного) индикатора, либо химическая адсорбция основания (кислоты).
Кислоты и основания Брэнстеда образуются в результате адсорбции молекулы воды или ее фрагментов на соответствующих центрах Льюиса, и на поверхности твердого оксида могут быть представлены гидроксильными группами трех типов: ОН‾, ОН∙, ОН+, а также в разной мере депротонированными молекулами воды, связанными с основными и кислотными центрами Льюиса, соответственно по кислотному или основному механизмам.
МеОδ–…Н–ОН (a)
Меδ+…О–Н (б) \ Н |
Рис. 1. Депротонированные молекулы воды, связанные с основными и кислотными центрами Льюиса, по кислотному (а) и основному (б) механизмам |
На рисунке 2 приведена формальная схема частично дегидратированной гипотетической поверхности твердого оксида, где тип и сила центров выражены в единицах рКа. Для простоты изложения и понимания подхода здесь не рассматривается процесс физической адсорбции воды.
Н Н Н-ОН Н \ / . Н │ Н О : │ О О . О2– О2– О │ │ : Э Э Э Э Э Э + Э+ |
||
Область льюисовских основных центров |
Область Бренстеда |
Область льюисовских кислотных центров |
Рис. 2. Формальная схема кислотно-основных центров реальной поверхности твердого тела |
||
Поверхность твердого оксида в условиях гидратации представляет собой набор «закрепленных» кислот и оснований, количество и сила которых могут быть зафиксированы с помощью адсорбции молекул-зондов (кислот и оснований) и выражена через их рКа, т.е. в единицах шкалы рН.
Большинство процессов, протекающих с участием поверхности твердых веществ, носят локальный характер и во многом определяются энергетическими параметрами конкретных активных центров. В связи с этим особую важность приобретает исследование спектра распределения центров адсорбции по кислотно-основному типу и силе и характере его изменения в зависимости от тех или иных условий.
Существует несколько различных методов исследования поверхностной кислотности твердых веществ, однако, большинство их них связано в той или иной форме с адсорбцией (ионный обмен, потенциометрическое титрование, адсорбция газообразных реагентов, методы оптической и ИК-спектроскопии, индикаторный метод). Наиболее информативными в этом плане являются спектральные методы. К настоящему времени для количественного анализа твердой поверхности на содержание кислотно-основных центров с дифференциацией их по типу и силе наиболее разработан индикаторный метод в спектрофотометрическом варианте.
Согласно современным воззрениям, поверхность твердого вещества бифункциональна, поскольку представляет собой совокупность центров Льюиса и Брэнстеда как кислотного, так и основного типа. Принадлежность центров к льюисовскому (апротонному) типу определяется наличием акцепторного или донорного поверхностного состояния, локализованного на полностью координированных атомах элементов вещества на его поверхности, которому следует приписать соответствующее положение на шкале энергий.
Изменения в электронной системе поверхности образца под влиянием тех или иных факторов приводит к сдвигу в соответствующем направлении кислотноосновного равновесия, выражающегося в изменении констант диссоциации поверхностных центров. С уменьшением значений рКа растет кислотность поверхности по Бренстеду. При гидратации центры Льюиса переходят в соответствующие индивидуумы бренстедовского типа.
Большинство процессов, протекающих с участием поверхности твердых веществ, носят локальный характер и во многом определяются энергетическими параметрами конкретных активных центров. В связи с этим особую важность приобретает исследование спектра распределения центров адсорбции по кислотно-основному типу и силе и характере его изменения в зависимости от тех или иных условий.
Индикаторный метод
Индикаторный метод – наиболее старый и экспериментально простой способ измерения кислотности поверхности твердого тела. Он основан на том, что, адсорбируясь, индикатор может менять окраску, которая является мерой кислотности (основности) его поверхности.
Полное описание кислотно-основных свойств поверхности твердого вещества подразумевает определение концентрации и силы активных центров, т.е. получение их распределения с дифференциацией на кислоты и основания, а также установление величины ее функции кислотности (Но).
История метода начинается с того момента, когда Гаммет выдвинул представление о функции кислотности для характеристики водных растворов и предложил серию индикаторов, позволяющих контролировать различные среды в широком диапазоне рН. Уоллинг перенес понятие функции Гаммета на случай твердых поверхностей и впервые применил метод для исследования различных классов веществ, использовав для этих целей набор индикаторов Гаммета. Последователи Уоллинга расширили ассортимент реагентов и доступный для исследований диапазон Н0.
Изучение свойств и строения кислотно-основных индикаторов в водных растворах показывает, что, несмотря на сложность процессов, определяющих изменение окраски, их можно описать с помощью классического уравнения кислотно-основного равновесия:
HInd (окраска I)↔ Ind– + H+ (окраска II),
которое
характеризуется константой диссоциации:
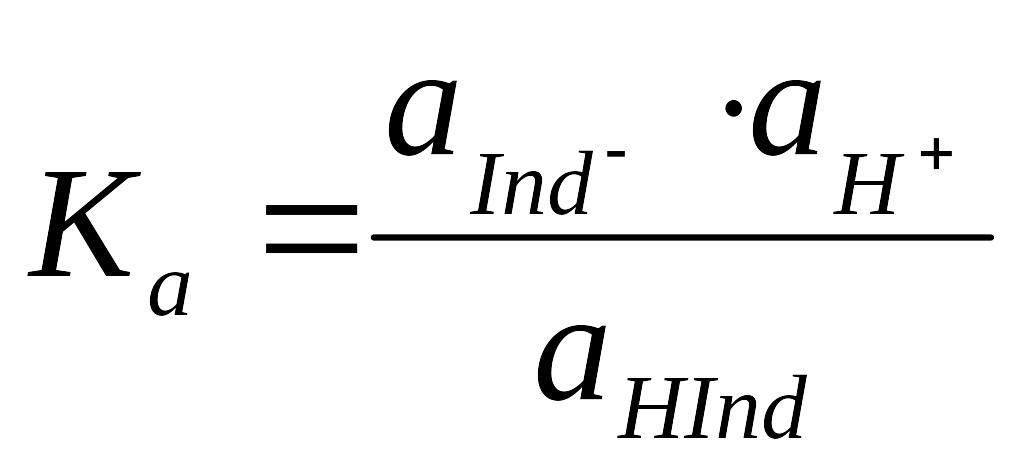 (1),
(1),
где a(H+), a(Ind–) , a(HInd) – термодинамические активности протона, основной и кислотной форм индикатора. Ka зависит от многих параметров – рН, ионная сила раствора, фазовое состояние исследуемых систем, природа растворителя..
Термодинамическая
активность:
![]() ,
(2)
,
(2)
где f – коэффициент активности иона, с – концентрация иона.
Подставляем
в уравнение для константы диссоциации
и преобразуем:
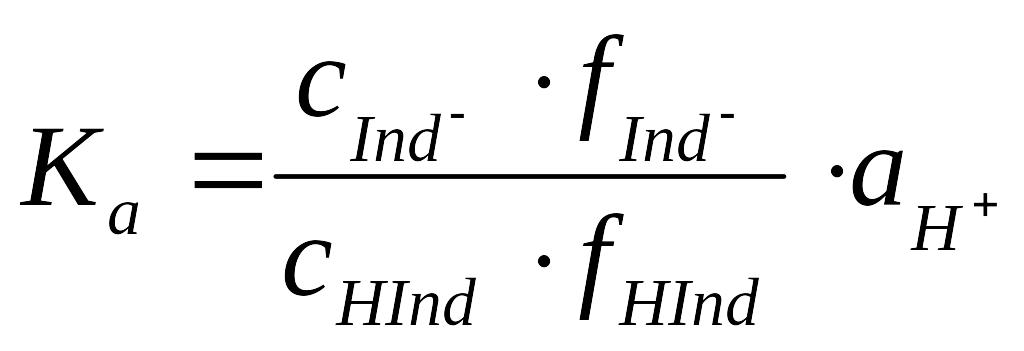 (3)
(3)
Соотношение
 называется
кислотностью среды.
называется
кислотностью среды.
При
логарифмировании выражения (3) получаем:
![]() ,
,
подставив
выражения для функции кислотности:
![]() и показателя кислотности среды
и показателя кислотности среды
![]() получим:
получим:
![]() ,
тогда
,
тогда
 (4)
(4)
Функция кислотности Н0 - есть мера кислотности среды, в качестве которой в данном случае выступает поверхность твердого вещества. Н0 можно определить по величине отношения концентраций основной и кислотной форм индикатора:
[Ind–]/[HInd], что может быть зафиксировано спектрофотометрически).
– показатель кислотности, характеризует кислотную силу индикаторов, являющихся слабыми кислотами или основаниями.
Величина силы кислоты или основания – понятие энергетическое и выражается через величины соответствующих констант диссоциации, являющихся мерой энергии химической связи и может быть выражена через изменение изобарно-изотермического потенциала (энергии Гиббса) ΔG = –RT ln KHInd. Величина ln KHInd отражает различия в энергиях связи между реагирующими компонентами, каковыми выступают центр адсорбции и молекула индикатора.
Согласно современной электронной теории коллективных взаимодействий вся химическая индивидуальность адсорбированной молекулы скрыта, “закодирована” в одном-единственном параметре – положении ее локального уровня, который с позиций теории кислот и оснований может быть охарактеризован через соответствующее значение рКа.
Появление окраски, соответствующее кислотной форме адсорбированного индикатора, указывает на то, что функция кислотности поверхности Н0 ниже значения рКа применяемого индикатора. Аналогично при адсорбции на твердом основании кислотного индикатора появляется окраска, характерная для его сопряженной основной формы. Это говорит о том, что на поверхности твердого тела имеются центры основной силы, достаточной для передачи пары электронов молекуле кислоты. Наблюдая за изменением цвета индикаторов в определенном интервале значений рКа , можно оценить кислотно-основную силу поверхности твердого вещества. Промежуточная окраска адсорбированного на поверхности индикатора соответствует равному содержанию его кислотной и основной форм, т.е. [Ind–]/[HInd]=1. И тогда приближенно можно принять, что в момент изменения цвета Н0= рКа. Таким образом, подбирая и применяя большое число индикаторов с различными значениями рКа, можно охватить интервал твердых веществ с большим различием в функции кислотности, сравнительно просто оценить относительную силу кислотно-основных центров, присутствующих на поверхности, и, определив концентрацию, получить распределение их по типу и силе.
На рис. 3 приведены кривые распределения центров адсорбции (РЦА), построенные в координатах q pKax = f(pKax ), где q pKax – содержание активных центров, эквивалентное количеству адсорбированного индикатора данной кислотной силы (pKax).
|
Рис. 3. Распределение кислотно-основных центров на поверхности |

Общая картина спектров РЦА проявляется в достаточно четкой дифференциации полос адсорбции с максимумами разной интенсивности, отвечающими определенным значениям рКа. Наличие полос в кислотно-основных спектрах говорит о присутствии на поверхности различных энергетических групп центров, которые могут быть охарактеризованы интервальными значениями рКа.
Под влиянием тех или иных факторов или внешних воздействий (например, “дегидратация”, рис.3, кривая 1) энергетическое состояние поверхности изменяется. Это проявляется в изменении распределения центров адсорбции, выражающееся в сдвиге всего спектра РЦА или отдельных полос и вариации их интенсивности.
Поскольку Н0 поверхности является величиной среднестатитистической, она может быть приближенно из спектров РЦА по формуле:
Н0 = Σ pKax∙ qpKax/Σ qpKax (5)
Тестовые каталитические реакции.
При конверсии (превращении) метилбутинола распределение продуктов реакции полностью характеризует кислотные, основные и амфотерные центры, так как различные продукты могут образовываться при одной и той же температуре без всяких ограничений.
|
Рис. 4. Схема конверсии метилбутинола
|

Механизм трансформации метилбутинола в ацетон и ацетилен показан на рисунке 3.16. Ион кислорода поверхности катализатора атакует водород гидроксогруппы субстрата, превращая его в промежуточное соединение типа алкоголята. Расщепление связи С-С и перегруппировка связи С-О приводит к образованию ацетона и ацетилена. Химическая природа активных центров при этом остается неизменной к концу процесса.
|
Рис. 5. Механизм превращения метилбутинола на основных центрах |

На кислотном центре поверхности метилбутинол формирует иное переходное состояние, в котором кислород алкоголята взаимодействует с водородом ОН-группы поверхности (рисунок 1.5). Затем реакция может проходить двумя различными путями. Первый вариант предполагает элиминирование молекулы воды, после которого восстанавливается кислотный центр и образуется 2-метил-1-бутен-3-ин. Для формирования 3-метил-2-бутен-1-аля первый этап аналогичен, но на следующем этапе происходит перемещение связи С-О от второго атома углерода к четвертому, образуется новый неустойчивый интермедиат, который подвергается кето-енольной перегруппировке. На последнем этапе восстанавливается кислотный центр и образуется альдегид 3-метил-2-бутен-1-аль.
|
Рис. 6. Механизм превращения метилбутинола на кислотных центрах |

На амфотерных центрах образуются 3-гидрокси-3-метил-2-бутанон и 3-метил-3-бутен-2-он. Предлагаются различные механизмы образования этих продуктов: возможно, эти вещества взаимопревращаются друг в друга при реакции гидратации-дегидратации. Вода присоединяется по тройной связи с образованием промежуточного енола, который превращается на основании кето-енольной таутомерии в 3-гидрокси-3-метил-2-бутанон, затем путем дегидроксилирования может образовываться 3-метил-3-бутен-2-он. Также для метилбутинола существует возможность присоединить слабосвязанный протон от кислотного центра, тогда возникает карбокатион, который переходит в более стабильное состояние, при отщеплении протона образуется 3-метил-3-бутен-2-он, а само отделение протона катализируется основным каталитическим центром.
|
Рис. 7. Механизм превращения метилбутинола на амфотерных центрах |

Еще одной реакцией, с помощью которой можно охарактеризовать кислотно-основные свойства поверхности это реакция Кнёвенагеля. Конденсация Кнёвенагеля – модифицированная альдольная конденсация. Различные типы альдольной конденсации и конденсации Кнёвенагеля используют в органическом синтезе, например, при производстве красителей, и гетерогенном катализе. По своей сути это конденсация альдегидов или кетонов с соединениями, содержащими активную метиленовую группу, в жидкой среде, в присутствии оснований с образованием производных этилена. В качестве исходных веществ используют большой спектр соединений.
|
Рис. 8. Схема реакции конденсация Кнёвенагеля, где R1 и R2 – могут быть CHO, COR, COOH, CN, NO2, SOR, SO2R и др. подобные. |

В большинстве работ каталитическая активность катализаторов в отношении реакции конденсации Кнёвенагеля в ее различных модификациях оценивается по выходу продукта за определенное время.
Выход продуктов каталитических реакций оценивается чаще всего с помощью газо-жидкостного хроматографа.
Ни один из трех описанных метода не дает разделения активных центов на центры Льюиса и Бренгстеда.