

Анемии вследствие нарушения кровообразования ( дизэритропоэтические анемии)
По патогенезу нарушения эритропоэза выделяют следующие их разновидности: 1) дисрегуляторная , обусловленная количественным и качественным нарушением синтеза эритропоэтина (снижением продукции их стимуляторов и увеличением образования их ингибиторов), синтеза и активности ферментов, участвующих в образовании порфирина, гема и/или глобина;
2) дефицитная, развивающаяся из-за недостатка в организме неорганических и органических веществ, крайне необходимых для синтеза полноценных эритроцитов; 3) гипопластическая или апластическая, возникающая в результате угнетения красного ростка костного мозга вследствие либо его разрушения, либо его сдавления или замещения другой тканью.
ДЕФИЦИТНЫЕ АНЕМИИ
К ним относятся: а) железодефицитные (частота их развития составляет 80-95 %); б) В12-дефицитные; в) фолиево-дефицитные; г) белково-дефицит- ные; д) смешанные.
Железодефицитные анемии
Следует отметить, что в норме из 10-15 мг железа, поступающего в сутки с пищей организмом утилизируется только 1,2-1,5 мг.
Этиология. Железодефицитные анемии возникают в результате:
1) снижения поступления железа с пищей;
уменьшения всасывания железа через слизистую тонких кишок (главным образом в дуоденуме и верхней части тощей кишки) в результате:
а) дефицита или отсутствия соляной и других (аскорбиновой, лимонной и др.) кислот, приводящих в норме к превращению (восстановлению) оксидного железа (Fe3+ ) в закисное железо (Fe2+), которое в виде ферритина (как и гемоглобин, и миглобин) у здорового человека хорошо всасывается через слизистую кишечника,
б) повреждения микро- и макроворсинок слизистой кишки;
3) расстройств транспорта вновь окисленного в плазме крови железа в комплексе с трансферритином (-глобулином), что наблюдается при гипотрансферритикемии, наличии антител к трансферритину;
нарушения (снижения) депонирования железа в печени, костном мозгу и мышцах (в норме составляющего около 20% от циркулирующего в крови количества);
уменьшения утилизации железа тканями, особенно костным мозгом, что отмечается при инфекциях, интоксикациях, гепатитах, циррозе печени и др. (в норме составляющего около 80% циркулирующего в крови количества железа);
резкого повышения потребления железа тканями (при беременности, лактации, быстром росте тела);
потери железа организмом (в котором ведущая роль принадлежит массивным острым или небольшим, но хроническим кровопотерям).
Патогенез данной анемии представлен на схеме 23-2. Экзогенный или эндогенный дефицит железа характеризуется снижением его содержания в сыворотке крови, костном мозге и истощением резервов железа, что проявляется исчезновением гемосидерина (темно-желтого железосодержащего пигмента) в макрофагах печени и селезенки, снижением содержания сидеробластов в костномозговой ткани. В результате уменьшается синтез гемоглобина и пролиферативная активность ядерных эритроидных клеток. Дефицит железа отражается также на активности тканевого дыхания вследствие снижения уровня железосодержащих клеточных ферментов (цитохрома С, цитохромоксидазы, сукцинатдегидрогеназы, пероксидазы).
Клиническая картина. Развивается гипохромная (ЦП 0,86), микроцитарная анемия. Появляется много патологически измененных форм (пойкилоцитов и анизоцитов).
Развивающаяся не только гемическая, но и тканевая гипоксия при железодефицитных анемиях приводит к формированию атрофических и дистрофических процессов в тканях и органах. В наибольшей степени это проявляется в поражении слизистой пищеварительного тракта, тканей миокарда, печени и почек. Признаками дефицита железа в организме являются также специфические сидеропенические симптомы – сухость и вялость кожи, повышенная ломкость ногтей, выпадение волос, разрушение зубов, мышечная слабость, извращение вкуса, ночное недержание мочи.
После снижения запасов железа компенсаторно стимулируется как всасывание, так и синтез трансферрина, а также эритропоэз (увеличение числа более мелких эритроидных клеток в результате активизации продукции эритропоэтина).
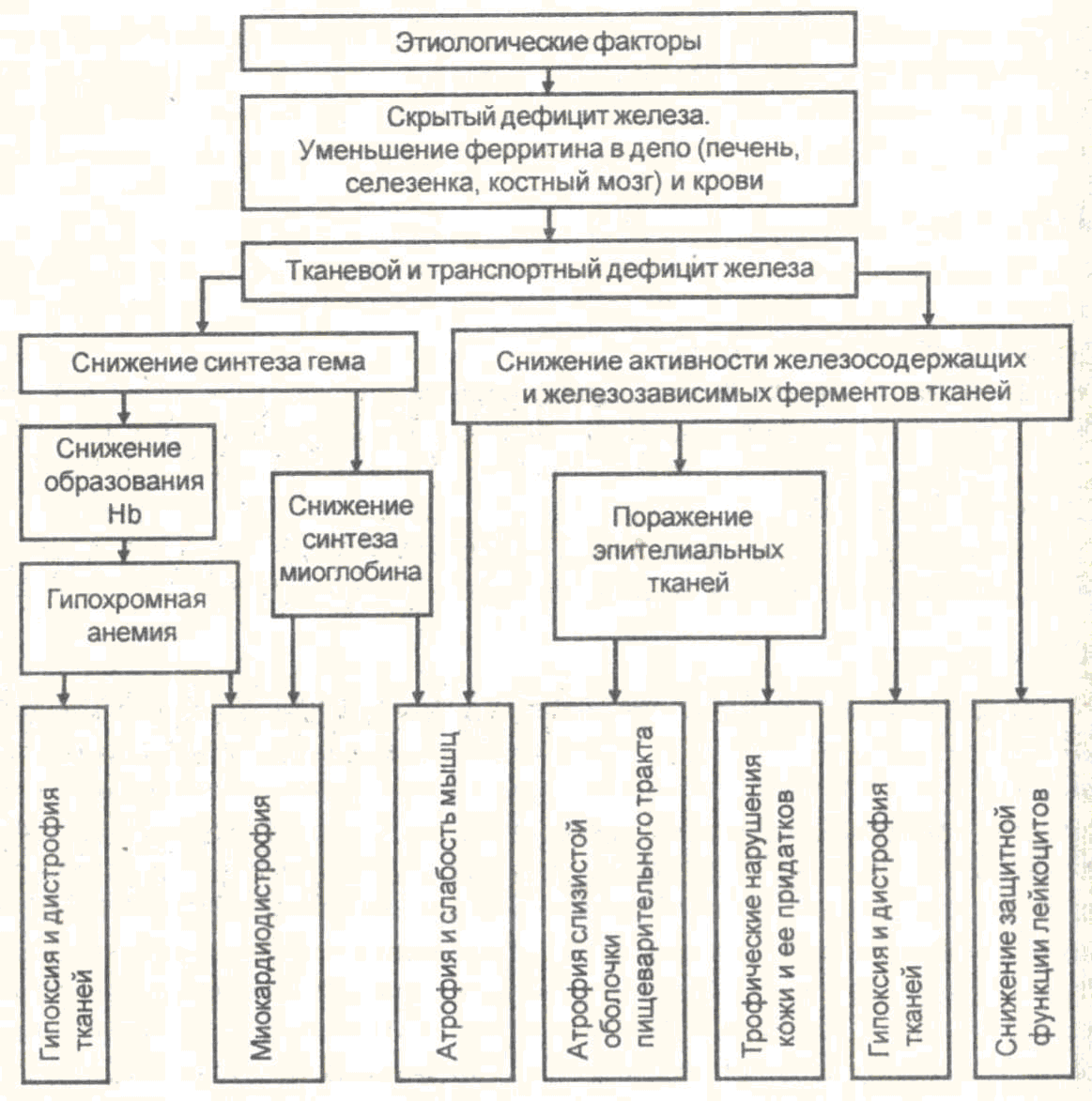
Схема 23-2. Патогенез железодефицитной анемии
(Окороков А.Н., 2001)
В12- и фолиеводефицитная анемия
Дефицит витамина В12 (внутреннего и внешнего фактора Кастла, цианокобаламина) и фолиевой (фолиновой) кислоты приводит к нарушению клеточного деления всех активно пролиферирующих клеток организма. Последнее связано со снижением синтеза пуриновых и пиримидиновых оснований, а следовательно - снижением синтеза нуклеиновых кислот (ДНК, РНК). На уровне системы крови развивается замена нормобластичеческого типа кроветворения мегалобластическим в сочетании с неэффективным миелопоэзом.
Этиология. Развитие абсолютного или относительного недостатка витамина В12 и фолиевой кислоты может определяться следующими факторами:
недостаточное поступление их с пищей (развивается только при продолжительном неполноценном питании, в том числе при вегетарианстве);
нарушение комплексирования витамина В12 с внутренним фактором Кастла (гастромукопротеином) в желудочно-кишечном тракте (особенно при дефиците внутреннего фактора Кастла, наблюдаемого при аутоиммунной агрессии, токсических, химических и термических поражениях, а также при врожденной недостаточности слизистой фундального отдела желудка или резекции желудка);
конкурентный расход витамина В12 (инвазия кишечника паразитами, повышенное потребление кишечными бактериями при их аномально интенсивном размножении);
нарушение чрезклеточного переноса витамина В12 (поражение слизистой тонких кишок, опухолевые процессы, интоксикации, дифиллоботриоз);
нарушение транспорта витамина В12 в крови и депонирования его в печени (при дефиците транспортного белка транскобаламина-2, относящегося к 1 -глобулинам; образование аутоантител, блокирование ферментных реакций, гепатиты, цирроз печени);
6) повышенная потребность в витамине В12 (быстрый рост организма, беременность, гипертиреоз, новообразования).
Патогенез. Дефицит витамина В12 и фолиевой кислоты приводит к снижению активности В12- зависимых энзимов, определяющих превращение фолиевой кислоты в ее коферментную форму тетрагидрофолиевую (фолиновую) кислоту, без которой невозможен синтез тимидина, входящего в состав ДНК. Это в основном и приводит к нарушению клеточного деления активно регенерирующих клеток кроветворной ткани. Развивается мегалобластический тип кроветворения, который характеризуется снижением числа митозов (нормо- бласт дает 3 митоза, мегалобласт – 1 митоз), удлинением времени митотического цикла, более ранним насыщением гемоглобином созревающих клеток эритроидного ряда, внутрикостномозговым разрушением мегалоцитов и укорочением (до 30-40 дней) продолжительности их жизни. Этиология и патогенез В12-дефицитной анемии представлены на схеме 23-3.
Клиническая картина. Формируется гиперхромная (ЦП1), мегалоцитарная анемия Аддисона-Бирмера, для которой характерны патологические включения в макроцитах (тельца Жолли, кольца Кебо, базофильная пунктация). При этом отмечается гиперсегментация ядер нейтрофилов и снижение в крови количества ретикулоцитов, лейкоцитов и тромбоцитов. Картина периферической крови представлена на схеме 23-5.
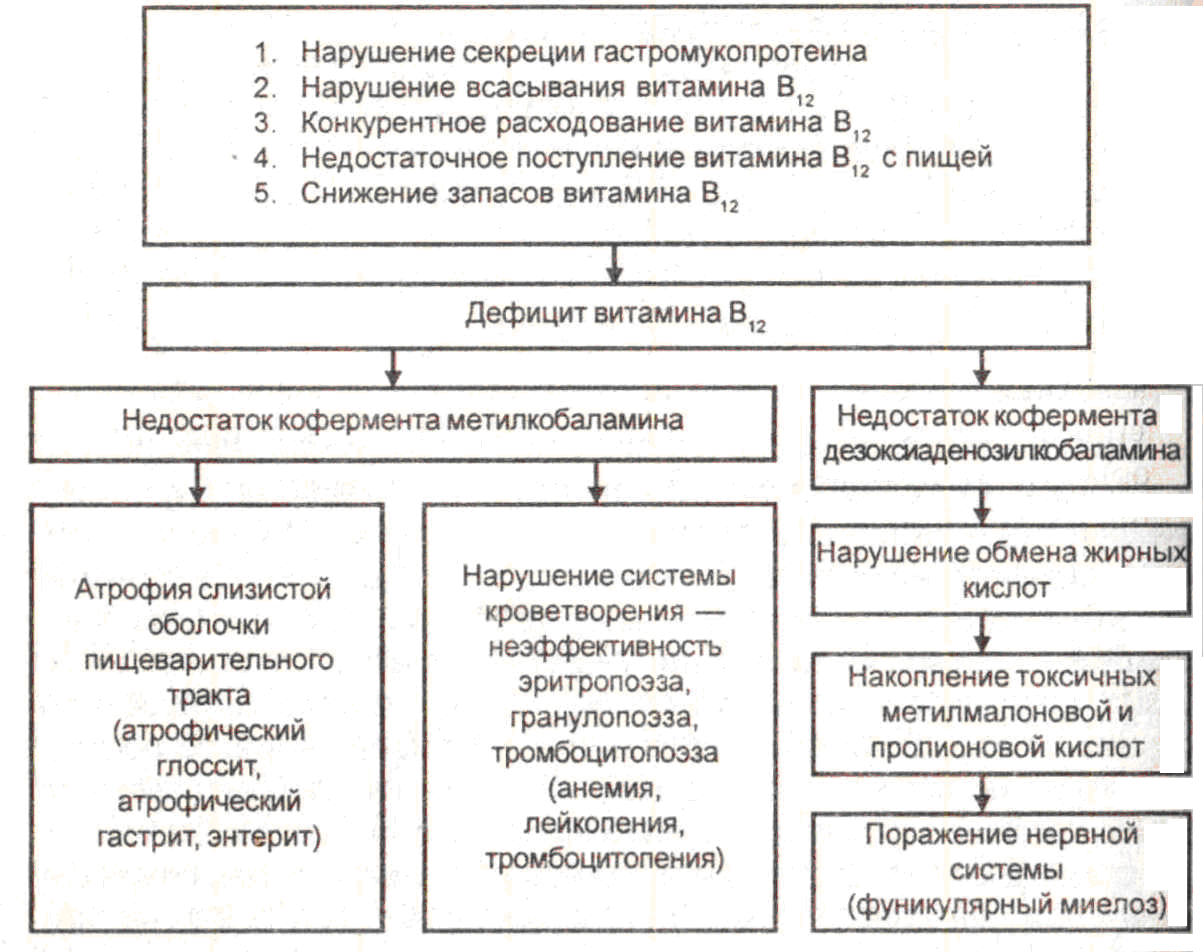
Схема 23-3. Этиология и патогенез В12-дефицитной анемии
(Окороков А.Н., 2001)
Изменения со стороны системы пищеварения проявляются в виде развития воспалительно-атрофических процессов в связи с нарушением регенерации эпителия слизистой пищевого канала (глоссит, стоматит, эзофагит, ахилический гастрит, энтерит).
Патология нервной системы (главным образом в виде дегенерации задних и боковых столбов спинного мозга – фуникулярного миелоза, а также поражений черепно-мозговых и периферических нервов) определяется тем, что при недостатке витамина В12 в тканях накапливаются пропионовая и метилмалоновая кислоты, которые являются токсичными для нервной ткани. При этом развиваются разнообразные неврологические симптомы – парестезии, дефицит моторики, расстройства глубокой чувствительности, болевой синдром и др.