

Генетичні дефекти ферментів синтезу сечовини
Існують спадкові ензимопатії, спричинені повним або частковим дефектом утворення в печінці окремих ферментів циклу сечовиноутворення. Найважчими клінічними проявами характеризуються порушення синтезу карбамоїлфосфатсинтетази та орнітинкарбамоїлтрансферази. Діти з такими генетичними дефектами страждають вираженою енцефалопатією, прояви якої дещо послаблюються в умовах повного виключення споживання харчових білків.
7. Загальні шляхи метаболізму вуглецевих скелетів амінокислот в організмі людини. Глюкогенні та кетогенні амінокислоти.
Двадцять L-амінокислот, що розрізняються за своєю хімічною структурою, біологічною роллю та особливостями метаболізму, входять до складу білків організму і присутні в клітинах та екстрацелюлярних просторах у вільному стані. Безазотисті скелети вільних амінокислот, які утворюються в результаті трансаміпувашія та дезамінування, - це метаболіти гліколізу, цитратного циклу, Р-окислення жирних кислот або речовини, що можуть перетворюватися в інтермедіати цих головних катаболічних шляхів організму.
Глюкогенні амінокислоти
L-Амінокислоти, що метаболізуються в циклі трикарбонових кислот, можуть включати свої вуглецеві скелети в молекули глюкози. Ці амінокислоти, використання яких у синтезі глюкози реалізується після їх входження в ЦТК через ацетил- КоА, а-кетоглутарат, сукциніл-КоА та фумарат, отримали назву глюкогенних амінокислот.
Кетогенні амінокислоти
Дві L-амінокислоти включаються в катаболізм тільки через ацетоацстил-КоА, якийу клітинах печінки може перетворюватися на кетонові тіла ацетоацетат та β-гідроксибутират. Це - кетогенні амінокислоти. Деякі амінокислоти віддають свої вуглецеві фрагменти на утворення як глюкози, так і кетонових тіл.
Кетогенез із амінокислот має особливо негативне значення при деяких порушеннях ферментних процесів, зокрема при некомпенсованому цукровому діабеті, у зв’язку з чим таким хворим рекомендується обмежувати надходження кетогенних амінокислот у складі продуктів харчування.
8. Біосинтез та біологічна роль креатину і креатин фосфату.
Креатин
- азотиста сполука, яка у вигляді
креатинфосфату має важливе значення в
енергозабезпеченні функції м’язів.
Біосинтез креатину відбувається за участю амінокислот гліцину, аргініну та метіоніну. Процес синтезу складається з двох стадій:
1-ша стадія - відбувається в нирках і полягає в утворенні глікоціаміну (гуанідинацетату) із аргініну та гліцину (фермент гліцинамідинотрансфераза).
2-а
стадія - відбувається в печінці, куди
тікоціамін надходить з 
током крові, і полягає в метилюванні глікоціаміну до креатину за
участю 8-аденозилметіоніну (фермент гуанідинацетатметилтрансфераза):
Фосфорилування
креатину при дії креатинфосфокінази
генерує креатинфосфат - джерело термінової
регенерації АТФ при м’язовому скороченні.
Незворотна
нефермснтативна
дегідратація і дефосфорилування
креатинфосфату
призводить до
утворення
ангідриду креатину - креатиніну.
У формі креатиніну з організму людини
виділяється із сечею значна частина
азоту амінокислот; у здорової людини
виділення креатиніну пропорційне масі
м’язових тканин і значно збільшується
за умов травматичних пошкоджень м’язів.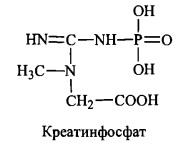
9. Глутатіон: будова, біосинтез та біологічні функції глутутіону.
Глутатіон - трипептид γ-глутамініл-цистеїннл-гліцин, що має у своєму складі вільну
сульфгідрильну групу:

Глутатіон міститься в клітинах тваринного організму у високій концентрації (близько
5 мМ). Глутатіон оборотно перетворюється з відновленої (Г-SН) до окисленої
(Г-S-S-Г) форми, відіграючи роль буфера SН-груп.
Біохімічна
функція глутатіону в організмі пов’язана
з відновленням і детоксикацією органічних
пероксидів - похідних пероксиду водню
НО-ОН, умолекулі якого один (гідропероксиди)
або обидва (алкілпероксиди) атоми водню
заміщені на алкільні радикали:
R-O-O-HR-O-O-R
При взаємодії глутатіону з гідропероксидом утворюються нешкідливі органічні спирти, що підлягають подальшому окисленню:
Реакція каталізується ферментом глутатіонпероксидазою, що містить в активному центрі атом селену (Sе).
Зворотне відновлення Г-SS-Г до Г-SH каталізується НАДФН-залежною глутатіонредуктазою.
10. Спеціалізовані шляхи метаболізму циклічних амінокислот - фенілаланіну та
тирозину.
Особливістю метаболізму в тваринних організмах циклічних амінокислот фенілаланіну та тирозину є утворення з них численних фізіологічно активних сполук гормональної та медіаторної дії, а саме: катехоламінів (адреналіну, норадреналіну), тиреоїдних гормонів, меланінів (рис. 18.7).
Шляхи метаболізму фенілаланіну:
1.Катаболічний шлях обміну полягає у втраті фенілаланіном аміногрупи (в реакції трансамінування) з утворенням фенілпірувату та кінцевого метаболіту фенілацетату, що екскретується з організму.
2. Шлях синтезу фізіологічно активних сполук починається з перетворення фенілаланіну на тирозин при дії ферменту фенілаланіпгідроксилази з подальшим перетворенням тирозину (див. нижче).
Шляхи метаболізму тирозину:
1. Катаболічний шлях обміну полягає у трансамінуванні тирозину і перетворенні його на р-оксифенілпіруват, який окислюється до гомогентизинової кислоти у складній реакції, коферменту роль у якій виконує аскорбінова кислота (вітамін С); подальші перетворення полягають в окисленні гомогентизату до фумарилацетоацетату (фермент оксидаза гомогентизинової кислоти) та розщепленні фумарилацетоацетату до фумарату та ацетоацетату.
2. Шлях синтезу катехоламінів та меланінів (пігментів шкіри). Шлях починається з окислення тирозину за участю специфічної гідроксилази до 3,4-діоксифенілаланіну (ДОФА), на рівні якого відбувається дивергенція двох обмінних шляхів: утворення катехоламінів (через декарбоксилування до дофаміну) та меланінів (через окислення
тирозиназою до дофахінону).
3. Шлях синтезу тиреоїдних гормонів-реалізується в клітинах щитовидної залози і полягає в утворенні йодованих тиронінів.
11. Спадкові ензимопатії обміну циклічних амінокислот: фенілкетонурія, альбінізм.
Фенілкетонурія - ензимопатія, спричинена генетичним дефектом синтезу фенілаланінгідроксилази. Внаслідок блокування утворення тирозину з фенілаланіну останній у збільшеній кількості надходить на шлях утворення фенілпірувату та фенілацетату, які в надмірних концентраціях накопичуються в організмі хворих. Концентрація фенілаланіну в крові хворих зростає в десятки разів, досягаючи 100-800 мг/л (норма -10-40 мг/л). Патологія проявляє себе ранніми порушеннями психічного розвитку дитини - фенілпіровиноградна олігофренія (oligophrenia phenylpyruvica).
Альбінізм - ензимопатія, біохімічною основою якої є спадкова недостатність ферменту тирозинази, що каталізує реакції, необхідні для утворення чорних пігментів меланінів. Відсутність меланінів у меланоцитах шкіри проявляється недостатньою (або відсутньою) пігментацією шкіри та волосся, підвищеною чутливістю шкіри до
сонячного світла, порушенням зору.