

Электронный спектр
Электрохимическая
ячейка
Электролитическая
ячейка
Эмиссионные спектры
Распределение эмитируемых электронов с поверхности образца по кинетическим энергиям под воздействием пучка рентгеновского излучения или электронов; эмиссия электронов происходит с внутренних электронных уровней атома и содержит небольшое число характеристических линий
Электрохимическая ячейка состоит из пары электродов, погруженных в раствор электролита: электроды могут быть помещены в один раствор (ячейка без жидкостного соединения) или в разные растворы, которые контактируют через пористую перегородку или через солевой мостик.
Электролитическая ячейка - это электрохимическая ячейка, во внешней цепи которой протекает электрический ток за счет источника постоянного напряжения, следовательно, она является потребителем внешней энергии Спектры испускания, возникающие при термическом
возбуждении, называют эмиссионными спектрами.
3.4. Технические и программные средства обеспечения дисциплины
1.По разделу 3 используются компьютерные программы DEMO по темам:
1.1.Атомно-абсорбционная спектроскопия (AAS);
1.2.Атомно-эмиссионная спектроскопия с индуктивно-связанной плазмой (ICP).
2.Демонстрационное приложение к курсу «Современная аналитическая аппаратура».
3.Средства обеспечения освоения дисциплины (ресурсы Internet)
1)http://www.bookam.net
2)http://www.nehudlit.ru
3 ) http://anchem.ru/forum/
4)http://anchem.ru/literature/books/
5)www.labware.ru
6)www.rusanalytchem.org.
92
3.5.Методические указания к выполнению лабораторных работ
Техника безопасности при выполнении лабораторных работ
Организация безопасной работы в лабораториях кафедры производится в соответствии с требованиями ГОСТ 12.1.019-79 «Электробезопасность. Общие требования», ГОСТ 12.1.030 – 81 «Электробезопасность. Защитные заземления. Зануление».
К работе допускаются студенты, изучившие инструкцию по технике безопасности и расписавшиеся в соответствующем журнале.
Запрещается проводить работу без задания преподавателя.
Соблюдение правил техники безопасности является обязательным. Студенты, нарушившие их, от работы отстраняются.
Дополнительные правила техники безопасности, касающиеся выполнения конкретной работы, указаны в методических указаниях к этой работе.
ОБЩИЕ УКАЗАНИЯ
Внастоящем указании рассматриваются методики выполнения лабораторных работ по химическим (титриметрия) и инструментальным методам анализа (оптическим, электрохимическим и рентгенофлуоресцентным). Все они используются в практике сервисной аналитической службы металлургических
имашиностроительных предприятий, обеспечивающей конкретный анализ определенных объектов с использованием методов, рекомендуемых аналитической химией. Выполнение представленных работ позволяет на модельных системах освоить принципы построения аналитической методики и обработки результатов анализа в химическом и инструментальном анализе, а также ознакомиться с возможностями использования аналитической аппаратуры для анализа входного сырья, продуктов технологического процесса и конечной продукции.
Всоответствии с рабочей программой дисциплины из приведенного перечня работ студент очной формы обучения выполняет все лабораторные работы, студенты очно-заочной формы обучения выполняет по указанию преподавателя одну работу по титриметрическому анализу (№ 1 или № 2) и три работы по физико-химическим методам анализа (с № 3 по № 10). Студенты заочной формы обучения выполняют по указанию преподавателя одну работу по тит-
93
риметрическому анализу и одну работу по физико-химическим методам анализа (с № 3 по № 10).
Перед началом выполнения каждой работы необходимо ознакомиться с соответствующей теоретической частью, приведенной в данном сборнике и в рекомендованной литературе для каждой группы лабораторных работ. Выполненные работы оформляются в отдельной ученической тетради в соответствии с приведенными рекомендациями к каждой работе. Калибровочные графики необходимо строить на миллиметровой бумаге. Для подготовки к защите выполненной работы следует ответить на вопросы, приведенные к каждой работе.
ВВЕДЕНИЕ
Для определения химического состава входного сырья, продуктов технологического процесса и конечной продукции металлургических и машиностроительных производств применяют химические, физико-химические и физические методы анализа. Выбор метода анализа основывается на требованиях к необходимой точности, чувствительности, скорости определения химического состава анализируемого объекта, а также стоимости анализа.
Основная часть производственных аналитических задач решается в настоящее время с помощью инструментальных (физических и физикохимических) методов анализа. В первую очередь, это применение оптических и рентгенофлуоресцентных квантометров для проведения маркировочного анализа готовой продукции, а также анализа химического состава металла и шлаков по ходу плавки в сталеплавильных цехах металлургических и машиностроительных заводов. В дополнение к ним для решения отдельных задач используются, например, кулонометрические анализаторы на углерод и серу, фотометрические, химические и другие методы.
Маркировочный анализ позволяет оценить соответствие состава исходного сырья, продуктов производства требуемым нормативам. Поэтому определение состава должно быть проведено с высокой точностью.
При возникновении необходимости проверки результатов маркировочного анализа проводят контрольный анализ, для которого используют, как правило, те же методы, что и для маркировочного, но выполняется он несколькими специалистами.
94
Химические методы анализа используются для определения высоких содержаний элементов, для установления состава стандартных образцов, для проведения арбитражных анализов.
Таким образом, аналитическая сервисная служба современных металлургических и машиностроительных предприятий для полного осуществления своих функций должна располагать как можно более полным арсеналом физических, химических и физико-химических методов анализа.
I.ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
(ЛАБОРАТОРНЫЕ РАБОТЫ № 1 И 2)
Химические методы анализа включают титриметрические и гравиметрические методы.
Вданном лабораторном практикуме рассматриваются только титриметрические методы.
Титриметрия включает кислотно-основные, окислительно-восстано- вительные, осадительные и комплексонометрические методы анализа.
Титрование может проводиться прямым, обратным и заместительным способом [1, 4].
1.ТИТРИМЕТРИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Вкачестве примера для ознакомления с основными этапами построения методики титриметричеких определений, освоения приемов титрования, принципов расчетов результатов анализа в титриметрии рассмотрим прямое комплексонометрическое титрование и заместительное окислительно-восстанови- тельное титрование (иодометрия).
95
ЛАБОРАТОРНАЯ РАБОТА №1 КОМПЛЕКСОНОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ
ОКСИДОВ КАЛЬЦИЯ И МАГНИЯ В ШЛАКАХ
1.ЦЕЛЬ РАБОТЫ
1.Ознакомление с приемами комплексонометрического титрования, применяемыми для определения суммарного и раздельного содержания нескольких компонентов в анализируемом растворе, на примере определения содержания оксидов кальция и магния в шлаках.
2.Определение содержания оксидов кальция и магния в шлаках.
2.ОСНОВНЫЕ ТЕОРЕТИЧЕСКИЕ ПОЛОЖЕНИЯ
При получении железных сплавов в качестве вспомогательных материалов могут быть использованы шлаки, в состав которых входят оксиды металлов и неметаллов и другие компоненты. Оксиды в составе шлаков подразделяются на основные (CaO, MgO, FeO, MnO, Na2 O, K2 O); кислотные (SiO2, P2O5, TiO2 , V2O5); амфотерные (Al2O3 , Cr2O3, V2O3). Проводимый в ходе металлургического процесса анализ шлаков позволяет регулировать режим ведения плавки с целью получения металла заданного качества. Обычно в шлаках определяют содержание оксидов кремния, кальция, магния, железа, серы, фосфора.
Анализ шлаков можно провести методом атомно-эмиссионной спектроскопии (квантометрически), но это связано с рядом трудностей, обусловленных большой сложностью химического состава шлаков (см. работу 7). Можно использовать для анализа шлаков рентгеновские квантометры, позволяющие проводить анализ с большой скоростью и точностью, сопоставимой с химическими методами. В случаях, когда эти методы нецелесообразны из-за высокой стоимости, используются химические методы, прежде всего, титриметрия.
В данной работе рассматривается определение содержания кальция и магния в шлаках прямым комплексонометрическим титрованием.
Рабочим раствором (титрантом) в комплексонометрии является динатриевая соль этилендиаминтетрауксусной кислоты (комплексон III, трилон Б, ЭДТА):
96
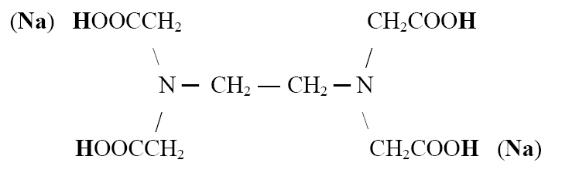
Сокращенное обозначение самой кислоты H4Y, а ее динатриевой соли - Na2 [H2Y].
Практически все катионы образуют очень устойчивые комплексы с ЭДТА, за исключением комплексов щелочных металлов. Независимо от степени окисления иона металла образование комплекса с ЭДТА протекает в соотношении 1:1. Степень связывания иона металла в комплекс с ЭДТА, а значит возможность его количественного титриметрического определения зависит от рН раствора и от величины константы устойчивости комплекса. В щелочной среде, где концентрация ионов-комплесообразователей Y4- наибольшая, практически все ионы металлов образуют комплексы, включая малоустойчивые комплексы ионов кальция и магния (соответствующие значения констант устойчивости 5·1010 и 4,9·108). Для создания щелочной среды используется аммонийная буферная смесь, поддерживающая постоянное значение рН в растворе около 10. Поскольку константы устойчивости комплексов кальция и магния с ЭДТА отличаются только на два порядка, оба иона титруются одновременно.
Все комплексы металлов с ЭДТА бесцветны, поэтому для установления конечной точки титрования используют индикаторы, которые называют металлиндикаторы. Они представляют собой органические многоосновные кислоты HnIn, собственная окраска которых резко отличается от окраски комплекса с металлами.
Для одновременного определения кальция и магния в качестве индикатора используется эриохромовый черный Т, образующий с ионами кальция и магния растворимые комплексы красного цвета с константами устойчивости, равными соответственно 2,6 106 и 1 107. Сам он имеет голубую окраску, а его комплексы с металлами окрашены в красный цвет. Значения констант устойчивости комплексов данных металлов с ЭДТА на порядок выше, поэтому в процессе титрования комплексы с индикатором разрушаются и образуются более проч-
97
ные комплексы ионов металлов с комплексоном III. Реакции, протекающие в растворе, схематически можно представить следующим образом:
Ca2+ + HIn2- = CaI n- + H+; Mg2+ + HIn2- = MgIn- + H+;
красный красный
CaIn- + Na2[H2Y] = Na2[CaY] + HIn2- + H+
красный бесцветн. бесцветн. синий
MgIn- + Na2[H2Y] = Na2[MgY] + HIn2- + H+
красный бесцветн. бесцветн. синий
В соответствии с этим в процессе титрования окраска раствора меняется от красной через фиолетовую в чисто синюю.
По результату титрования определяют суммарное содержание кальция и магния.
При необходимости определения каждого элемента в отдельности сначала определяют содержание кальция, титруя его после осаждения магния действием гидроксида натрия с использованием мурексида в качестве индикатора. Затем определяют суммарное содержание обоих компонентов в присутствии индикатора эриохрома черного Т. Содержание магния определяют по разности между результатами этих определений.
3.МЕТОДИКА ВЫПОЛНЕНИЯ РАБОТЫ
1.Определение содержания оксида кальция титрованием раствором ЭДТА после предварительного осаждения гидроксида магния действием гидроксида натрия с использованием мурексида в качестве индикатора.
2.Совместное титрование оксидов кальция и магния в приготовленном растворе шлака стандартным раствором ЭДТА в присутствии аммонийной буферной смеси с использованием эриохрома черного Т в качестве индикатора.
3.Определение содержания оксида магния по разности результатов титрования обоих компонентов и отдельного титрования кальция.
4.Расчет массовой доли оксидов кальция и магния в шлаке.
98

1. Определение содержания оксида кальция
Методика определения
Заполняют бюретку рабочим раствором ЭДТА. Получают у преподава-
теля в мерную колбу вместимостью 100 мл анализируемый объем Vан при-
готовленного раствора шлака, разбавляют до метки дистиллированной водой и тщательно перемешивают.
Приготовление раствора шлака. На аналитических весах берут навеску шлака mшлака ( 1,5 г) с точностью до четвертой значащей цифры, растворяют в хлороводородной кислоте, отделяют кремневую кислоту, гидроксиды железа, марганца, алюминия и переносят в мерную колбу на 250 мл и доводят до метки дистиллированной водой. Именно из этого раствора получают у преподава-
теля анализируемый объем Vан .(20…25 мл).
При растворении навески протекают следующие реакции:
(CaO)2 SiO2 + 4 HCl + (n - 1) H2O = H2SiO3 n H2O + 2 CaCl2 CaO Fe2O3 + 8 HCl = CaCl2 + 2 FeCl3 + 4 H2O;
при отделении мешающих элементов -
H2SiO3 n H2O H2SiO3 + n H2O, Fe3+ + 3NH4OH = Fe(OH)3 + 3NH4+
Mn(OH)2 + (NH4)2S2O8 + 2NH4OH = MnO2 +2 H2O + 2 (NH4)2 SO4.
Пипеткой отбирают из мерной колбы 4 аликвотные (равнодольные) части по 10 мл и переносят их в конические колбы для титрования. Добавляют в каждую по 5 мл раствора гидроксида натрия с концентрацией с(NaOH) = 2 моль/л, перемешивают и вносят небольшое количество сухого индикатора мурексида. Полученный мутный раствор титруют раствором ЭДТА с концентрацией с(1/2 ЭДТА) = 0,05 моль/л до перехода розовой окраски в сине-фиолетовую. Повторяют титрование 3-4 раза, результаты титрования записывают в рабочий журнал по форме 1:
Форма 1
Результаты титрования кальция раствором ЭДТА
Показания шкалы бюретки
h1 |
h2 |
h3 |
h4 |
hср |
|
|
|
|
|
99

Расчет содержания оксида кальция в анализируемом растворе
По усредненному значению показаний шкалы бюретки hср1 рассчитывают средний объем рабочего раствора V1(ЭДТА), необходимый для определения кальция во всем объеме приготовленного раствора (250 мл):
V1 (ЭДТА) = ( hср 100)/10 Vан. 250.
Массу оксидов кальция в приготовленном растворе шлака рассчитывают по формуле:
m(CaO)= [с(1/2 ЭДТА) V1(ЭДТА) M(1/2 CaO)] / 1000.
2. Совместное титрование оксидов кальция и магния
Методика определения
Пипеткой отбирают из мерной колбы еще 4 аликвотные части по 10 мл и переносят их в конические колбы для титрования. Добавляют в каждую колбу по 15 мл аммонийной буферной смеси, 50 мл дистиллированной воды и небольшое количество сухого индикатора эриохрома черного Т до получения малиновой окраски раствора. Титруют последовательно каждую колбу рабочим раствором ЭДТА до перехода малиновой окраски раствора через фиолетовую в ярко-голубую. Отмечают показания шкалы бюретки, эквивалентные суммарному содержанию оксидов кальция и магния в титруемой аликвотной части раствора, и записывают их в таблицу по форме 2.
Форма 2
Результаты совместного титрования кальция и магния раствором ЭДТА
Показания шкалы бюретки
H1 |
h2 |
h3 |
h4 |
hср |
|
|
|
|
|
Расчет объема ЭДТА, эквивалентного суммарному содержанию оксидов кальция и магния
По усредненному значению показаний шкалы бюретки hср2 рассчитывают средний объем рабочего раствора V2(ЭДТА), необходимый для определения оксидов кальция и магния во всем объеме приготовленного раствора шлака (250 мл):
V2 (ЭДТА) = (hср2 100)/10 Vан. 250.
100
3. Определение содержания оксида магния
По разнице объемов V2(ЭДТА) и V1(ЭДТА) находят объем V3(ЭДТА), эквивалентный содержанию оксида магния в приготовленном растворе шлака:
V3(ЭДТА) = V2(ЭДТА) - V1(ЭДТА).
Массу оксидов магния в растворе шлака рассчитывают по формуле: m(MgO)= [с(1/2 ЭДТА) V3(ЭДТА) M(1/2 MgO)] / 1000.
4. Расчет массовой доли оксидов кальция и магния в шлаке
Для вычисления заданных величин необходимо знать точную величину навески шлака mшлака. Тогда массовые доли обоих оксидов вычисляются на основе формул:
(CaO)= [m(CaO)/ mшлака] / 100(MgO)= [m(MgO)/ mшлака] / 100
4. СОДЕРЖАНИЕ ОТЧЕТА
Отчет о работе должен содержать:
1.Цель работы.
2.Краткое описание методики определения и уравнения химических реакций, протекающих в процессе титрования в схематической форме.
3.Результаты титрования в таблице по форме 1 и расчет содержания оксида кальция в приготовленном растворе шлака.
4.Результаты титрования в таблице по форме 2 и расчет содержания оксида магния в приготовленном растворе шлака.
5.Расчет массовой доли оксидов кальция и магния в шлаке.
Литература: [1],кн. 1, с. 189...204; [4],с. 23…24.
Вопросы для подготовки к защите лабораторной работы
1.Перечислите основные методы титриметрического анализа и виды титрования.
2.Запишите реакции, протекающие при анализе шлака комплексонометрическим методом.
3.Что показывает титр по определяемому веществу?
4.Составьте равенство количества эквивалентов для прямого титрования на примере определения кальция в шлаках.
101

ЛАБОРАТОРНАЯ РАБОТА №2 ИОДОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ МЕДИ В СПЛАВАХ
1.ЦЕЛЬ РАБОТЫ
1.Ознакомление с приемами окислительно-восстановительного титрования на примере заместительного иодометрического определения содержания меди в сплавах.
2.Определение содержания меди в сплаве.
2.ОСНОВНЫЕ ТЕОРЕТИЧЕСКИЕ ПОЛОЖЕНИЯ
Иодометрическое титрование относится к группе окислительно-
восстановительных методов. Оно может проводиться прямым, обратным и заместительным способом.
Для определения меди используется заместительное титрование: к определяемому окислителю (Сu2+) прибавляется избыток иодида калия, медь (II) восстанавливается до меди (I) с образованием нерастворимого осадка иодида меди CuI. Выделившийся свободный иод оттитровывается рабочим раствором тиосульфата натрия Na2S2O3 :
Cu2 I e CuI 2
2I 2e I2
2 Cu2+ + 4 I- = 2 CuI + I2
I2 + 2e- = 2 I-
2 S2O32- - 2e- = S4O62-
2 S2O32- + I2 = 2 I- + S4O62-
Расчет строится на равенстве количества эквивалентов определяемого вещества и рабочего раствора
n(Cu) = n(Na2S2O3).
При определении содержания меди в сплавах, рудах, высокотемпературных сверхпроводниках иодометрия не уступает по точности электрохимическому методу.
102
Из элементов, обычно сопутствующих меди в природных соединениях и промышленных сплавах, мешают ее иодометрическому определению железо, мышьяк и сурьма. Влияние железа устраняют связыванием в комплекс фторидили пирофосфат-ионами. Поскольку комплексные соединения этих ионов с железом (III) более устойчивы, чем с железом (II), то потенциал этой системы падает до значения, при котором окисление иодид-иона становится уже невозможным. Мышьяк (III) и сурьма (III) окисляются свободным иодом. Реакция протекает количественно при рН 3,5, хотя равновесие устанавливается медленно. Поэтому в процессе растворения пробы мешающее влияние мышьяка и сурьмы устраняют переведением в состояние окисления +5. Обычно с этой целью для растворения сплавов и руд используется горячая концентрированная азотная кислота. Окисление иодид-ионов мышьяком и сурьмой в степени окисления +5 происходит только в сильно кислой среде. При рН>3 влияние этих элементов можно устранить. Но при рН>4 медь (II) не полностью окисляет ио- дид-ионы. Поэтому в присутствии мышьяка и сурьмы определение меди можно вести при рН в интервале 3 - 4.
3. ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Выполнение работы включает следующие операции:
1.Иодометрическое определение содержания меди в анализируемом растворе сплава.
2.Расчет массовой доли меди в анализируемом сплаве.
3.Расчет абсолютной и относительной погрешности определения.
Методика определения
Получают у преподавателя определенный объем приготовленного раствора сплава меди в мерную колбу вместимостью 50 - 100 мл, доливают до метки дистиллированной водой, перемешивают и, промыв пипетку небольшим количеством раствора, отбирают ею 4 аликвотные части в колбы для титрования. Добавляют в каждую колбу 2 г иодида калия, по 5 мл 1 М раствора серной кислоты и сразу же титруют рабочим раствором тиосульфата натрия почти до полного исчезновения окраски иода. Когда раствор над осадком станет соло- менно-желтым, добавляют в колбу 3 мл крахмала и продолжают титрование до обесцвечивания синего раствора от одной избыточной капли тиосульфата. Показания шкалы бюретки записывают в рабочий журнал в таблицу по форме 3.
103
Последовательно оттитровывают три оставшиеся колбы и результаты титрования записывают в журнал.
Форма 3
Результаты титрования анализируемого раствора стандартизированным раствором тиосульфата натрия
|
|
Показания шкалы бюретки |
|
|
||
|
|
|
|
|
|
|
h1 |
h2 |
|
h3 |
|
h4 |
hср |
|
|
|
|
|
|
|
Расчет содержания меди в анализируемом растворе
По усредненным данным показаний шкалы бюретки рассчитывают объем тиосульфата натрия V1(Na2S2O3), пошедший на титрование выделенного иода. При этом необходимо учесть вместимость мерной колбы Vк и объем аликвотной части 10 мл:
V1(Na2S2O3) = (hср Vк) / 10.
Учитывая общий объем приготовленного раствора сплава 250 мл, объем раствора, выданный для анализа Vан, вычисляем объем тиосульфата V(Na2S2O3), эквивалентный всему количеству меди в анализируемом сплаве:
V(Na2S2O3)= V1(Na2S2O3) 250 Vан .
По значению известной величины молярной концентрации эквивалента тиосульфата натрия с(Na2S2O3) рассчитываем титр тиосульфата по меди:
T(Na2S2O3 / Cu) = с(Na2S2O3) 1000 М (Cu).
Теперь рассчитываем содержание меди в анализируемом растворе m(Cu):
m(Cu) = T(Na2S2O3 /Cu) V(Na2S2O3)
имассовую долю меди в сплаве
=[ m(Cu)/ mсплава ]/100 .
104
Расчет абсолютной и относительной погрешности определения
По данным контрольного (истинного) количества меди в анализируемом растворе m(Cu)ист, полученного у преподавателя, рассчитайте абсолютную и относительную D погрешности определения:
= m(Cu)ист - m(Cu)
D= [ / m(Cu)ист] 100% .
4.СОДЕРЖАНИЕ ОТЧЕТА
Отчет о работе должен содержать:
1.Цель работы.
2.Краткое описание методики определения и уравнения реакций в элек- тронно-ионной, ионной и молекулярной формах.
3.Факторы эквивалентности и молярные массы эквивалентов веществ, участвующих в реакции.
4.Результаты титрования в таблице по форме 3 .
5.Расчеты объема тиосульфата натрия, пошедшего на титрование выделенного иода.
6.Вычисление содержания меди в анализируемом растворе.
7.Расчет абсолютной и относительной погрешностей определения.
Литература: [1], кн.1, с.225…238; [2], кн.2, с.94…96.
Вопросы для подготовки к защите лабораторной работы
1.Перечислите основные методы титриметрического анализа.
2.Перечислите основные приемы титриметрического анализа.
3.Какой раствор называют титрантом или рабочим раствором? Чем он отличается от других растворов?
4.В чем сущность прямого титрования и титрования по замещению?
5.Что называют эквивалентом и молярной массой эквивалента?
6.Составьте равенства количества эквивалентов для прямого титрования на примере определения кальция в шлаках комплексонометрическим титрованием и титрования по замещению на примере определения меди в сплавах иодометрическим методом.
105
II. ФИЗИЧЕСКИЕ И ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
1. СПЕКТРОСКОПИЧЕСКИЕ ОПТИЧЕСКИЕ МЕТОДЫ АНАЛИЗА (ЛАБОРАТОРНЫЕ РАБОТЫ №3…8)
1.1. Спектр электромагнитного излучения
Методы прикладной спектроскопии основаны на изучении взаимодействия электромагнитного излучения с атомами или молекулами (ионами) исследуемого вещества. Для аналитических целей используется область от 106 до 1020 Гц. В эту область электромагнитного излучения входят радиоволны, микроволны, тепловое (инфракрасное), видимое, ультрафиолетовое
ирентгеновское излучения.
Врезультате взаимодействия возникает аналитический сигнал, содержащий информацию о свойствах исследуемого вещества: частота сигнала зависит от специфических свойств анализируемого соединения, то есть является основой для проведения качественного анализа, а интенсивность сигнала пропорциональна количеству вещества.
Атомная спектроскопия основана на поглощении или испускании рентгеновского, видимого или УФ-излучения. Она используется для качественного
иколичественного элементного анализа.
Рентгеновское излучение и поглощение обусловлено возбуждением внутренних, не участвующих в образовании связей электронов. Поэтому характер рентгеновского атомного спектра не зависит от химического состояния элемента в образце, а рентгеновский анализ проводится без разрушения образца.
Оптическая область включает инфракрасное, видимое и ультрафиолетовое излучения.
В видимой и УФ-области поглощение или излучение обусловлено возбуждением внешних валентных электронов. Поэтому чистый атомный спектр можно получить только после отделения анализируемого элемента от других связанных с ним элементов. С этой целью проводят атомизацию образца, в процессе которой молекулы распадаются на составные части и превращаются в атомы и ионы, существующие в газообразном состоянии.
Радиоскопические и рентгеновские методы анализа по технике резко отличаются от оптических.
106
1.2. Классификация спектроскопических оптических методов анализа
Оптическая область спектра включает область вакуумного ультрафиолета, область УФ-излучения, видимую и инфракрасную области спектра.
Методы анализа, основанные на исследовании взаимодействия электромагнитного излучения этой области с атомами и молекулами вещества, носят название оптических спектральных методов.
Оптический спектральный анализ включает абсорбционные методы, использующие спектры поглощения молекул (ионов) в В-, УФ- и ИК-областях, атомов в В- и УФ-областях и эмиссионные методы, использующие спектры излучения (эмиссии) атомов и ионов в УФ- и В-областях.
В абсорбционных методах исследуемое вещество вступает во взаимодействие с внешним электромагнитным излучением и поглощает (абсорбирует его), а в эмиссионных методах сам исследуемый образец под действием высокой температуры возбуждается и испускает электромагнитное излучение.
Молекулярный и атомный абсорбционный анализ в В- и УФ-областях используется только для количественного элементного анализа. Абсорбционные спектры в ИК-области при решении задач материаловедения используют для определения содержания газов в металлах.
С помощью атомно-эмиссионного анализа в В- и УФ-областях спектра решаются задачи установления качественного и количественного элементного состава пробы. Этот метод является ведущим при анализе сталей, сплавов,
сырья, полупродуктов и продуктов металлургического и машиностроительного производства.
Для него характерны высокая экспрессность (скорость выполнения анализа), высокая селективность, низкий предел обнаружения. Например, можно определять следовые количества веществ-загрязнителей при анализе чистых и сверхчистых материалов для атомной, полупроводниковой, электронной промышленности, в производстве чистых легких и цветных металлов. С помощью современных эмиссионных анализаторов можно одновременно определять содержание нескольких десятков элементов в анализируемом образце. При этом не требуется использовать длительные и трудоемкие предварительные операции химической пробоподготовки образца (сплавление, выщелачивание, удаление или маскирование мешающих компонентов).
Ограничения эмиссионного спектрального анализа возникают из-за невозможности определения высоких содержаний элементов, а также вследствие близости спектральных характеристик некоторых элементов, обуславливающих невозможность их определения при совместном содержании в пробе.
107
Эти задачи обычно решаются либо рентгеноспектральными методами (если нет финансовых ограничений), либо химическими или физикохимическими методами. Последние являются практически универсальными, но с экономической точки зрения, при большом пробопотоке их использование может быть только дополняющим к автоматическим многоэлементным анализаторам.
На современном рынке аналитического приборостроения для проведения всех видов спектрального анализа имеется большой выбор аппаратуры отечественных и зарубежных фирм. Среди последних наиболее широко представле-
на продукция следующих фирм: PERKIN – ELMER, VARIAN, HEWLETT PACKARD, HITACHI, SHIMADZU, INSTRUMENTATION LABORATORY, THERMO SPECTRONIK (USA), PYE-UNICAM, THERMO ELECTRON CORPORATION.
1.2.1. Абсорбционный спектральный анализ (лабораторные работы № 3…5)
Закон Бугера-Ламберта-Бера
Зависимость интенсивности поглощения монохроматического излучения от концентрации вещества и толщины поглощающего слоя выражается законом Бугера-Ламберта-Бера:
A = -lg I/I0 = ε lc,
где А - абсорбция или оптическая плотность (старое обозначение D); I0 - интенсивность падающего потока излучения; I -интенсивность потока излучения после прохождения поглощающего слоя; с- молярная концентрация; ε - молярный коэффициент поглощения.
Молярный коэффициент поглощения равен оптической плотности раствора при единичных значениях концентрации и толщины поглощающего слоя: с = 1моль/л и l = 1 см.
Молярный коэффициент поглощения не зависит от объема раствора, толщины слоя и интенсивности освещения. Он является качественной характеристикой вещества, характеризует его внутренние свойства и зависит только от природы анализируемого вещества и длины волны измерения. Поэтому его величина является объективной характеристикой возможной чувствительности фотометрического определения. Числовые значения величин молярного коэффициента поглощения в области максимума для различных поглощающих свет соединений одного и того же элемента могут отличаться на несколько порядков.
108
Поэтому измерение светопоглощения определяемого компонента проводят после перевода его в химическую форму с высоким значением величины молярного коэффициента поглощения. В видимой области спектра это яркоокрашенные комплексные соединения.
Оптическая плотность является аддитивной величиной, то есть для смесей нескольких поглощающих свет соединений, не взаимодействующих между собой, необходимо учитывать, что
Аобщ = А1 + A2 + A3 + ... + An .
Следовательно, для определения содержания заданного компонента
на основе метода молекулярно-абсорбционной спектроскопии необходимо предварительно удалить или замаскировать сопутствующие компоненты,
что, конечно, усложняет анализ и увеличивает время для его проведения. Однако, при этом метод становится практически универсальным, позволяя фактически решить любую задачу элементного анализа и являясь универсальным дополнением к эмиссионному спектральному анализу.
Спектр поглощения
Зависимость оптической плотности или молярного коэффициента поглощения от длины волны выражается кривой, называемой спектром поглощения. По оси абсцисс откладывают длину волны, а по оси ординат - оптическую плотность. В случае подчинения закону Бугера - Ламберта -Бера спектр сохраняет свой вид независимо от концентрации.
Аппаратура для молекулярно-абсорбционного анализа
Для измерения поглощения излучения в видимой области спектра используют фотоэлектроколориметры, фотометры и спектрофотометры в УФ-, В-
и ИКобластях. |
|
|
|
|
|
|
|
||
|
Вся аппаратура строится по общей схеме: |
|
|||||||
|
|
→ |
|
|
|
|
|
|
|
|
Источник |
Моно- |
Анализи- |
Приемник |
Измери- |
||||
|
сплошного |
→ |
хроматор |
→ |
руемый рас- |
→ |
излучения |
→ |
тельное |
|
излучения |
→ |
|
|
твор |
|
|
|
устройство |
|
|
|
|
|
|
|
|
|
|
Наиболее широко используются в практике работы заводских лабораторий - фотоэлектроколориметры ФЭК- М, ФЭК56, ФЭК-60, КФК – МП2, фотометры с дифракционной решеткой типа КФК 3.
109
Фотометрические реагенты
Почти всегда измерению светопоглощения предшествует перевод определяемого компонента в новую химическую форму, отличающуюся сильным поглощением. Это может быть окрашенное соединение, поглощающее свет в видимой области спектра, или соединения, поглощающие электромагнитное излучение в УФобласти. Разработка методики определения включает подбор такого соединения, выбор реагентов и условий для образования соединения, нахождение приемов устранения помех со стороны сопутствующих компонентов.
Если для анализа используется видимая область спектра, то методику называют фотоколориметрической или фотометрической.
В качестве фотометрических реагентов используют вещества различных классов, неорганические и органические. Поскольку большинство металлов и неметаллов способны к образованию окрашенных комплексов, то область применения фотометрии практически не ограничена ни по элементам, ни по объектам анализа.
Фотометрические методы могут быть использованы как для определения больших содержаний компонентов (20-30%), так и для определения микропримесей 10-4 – 10-5 %. Сравнительная простота оборудования, его невысокая стоимость, большое количество разработанных и аттестованных методик определения позволяют использовать этот метод практически во всех случаях, когда по каким-либо причинам невозможно использование экспрессных эмиссионных оптических или рентгенофлюоресцентных квантометров.
Примерами использования фотометрических методов в практике заводских лабораторий являются определения в сталях и сплавах фосфора в виде фосфорно-молибденового комплекса, кремния в виде синего кремнемолибденового комплекса, никеля в комплексе с диметилглиоксимом, хрома в комплексе с дифенилкарбазидом, молибдена на основе роданидного комплекса, ниобия, вольфрама, титана, меди, ванадия в виде различных окрашенных комплексов.
Поскольку измеряемое значение величины аналитического сигнала оптической плотности связано с концентрацией линейной зависимостью, то в зависимости от особенностей решаемой задачи для расчета концентрации могут быть использованы метод сравнения (для однократных определений), ме-
тод калибровочного графика (для серийных анализов), метод добавок (для определения содержания компонентов на границе чувствительности аппаратуры).
110
Для освоения принципов построения методики определения в фотометрии предлагаются три работы, в которых рассматриваются метод сравнения (работа №3), метод калибровочного графика (работа №4) и применение метода калибровочного графика для определения содержания никеля в сталях (работа №5).
ЛАБОРАТОРНАЯ РАБОТА №3 ФОТОКОЛОРИМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ХРОМА МЕТОДОМ СРАВНЕНИЯ
1.ЦЕЛЬ РАБОТЫ
1.Ознакомление с определением содержания вещества методом сравне-
ния.
2. Определение содержания хрома в исследуемом растворе.
2. ОСНОВНЫЕ ТЕОРЕТИЧЕСКИЕ ПОЛОЖЕНИЯ
Метод сравнения используется для однократных определений. Величины абсорбционности (оптических плотностей) эталонных и исследуемых растворов измеряют при одной и той же длине волны и толщине поглощающего слоя. Обычно приготавливают два - три эталонных раствора с тем, чтобы можно было определить среднее значение концентрации исследуемого раствора. Концентрации исследуемого и стандартных растворов должны быть близки по величине, чтобы избежать больших ошибок в измерении оптических плотностей.
Расчет содержания хрома производится по формуле:
сх = (сэ / Аэ)ср · Ах, (1)
где Аэ и Ах − значения абсорбционности (оптических плотностей) эталонного и исследуемого растворов; сэ и сх − значения величин концентраций эталонного и исследуемого растворов.
Метод изучается на примере определения концентрации хрома, содержащегося в исследуемом растворе в виде дихромат-ионов Сr2О72-. Максимум поглощения этого иона наблюдается при 340 нм. Поскольку в водных растворах, содержащих Сr(VI) существует равновесие:
Cr O |
2- + H |
O |
2CrO |
2- + 2H+ , |
|
2 |
7 |
2 |
|
|
4 |
определение проводится в кислой среде.
111
Краткое описание приборов
Измерение оптической плотности производится на фотоэлектроколориметре КФК. Прибор работает на основе двулучевой схемы, снабжен девятью светофильтрами с максимумами пропускания при следующих длинах волн
(нм): 315±5; 340±5; 400±5; 440±10; 490±10; 540±10; 582±10; 597±10; 630±10.
Принципиальные основы работы прибора изложены в теоретической части к разделу. Подробные правила проведения измерений прилагаются к прибору.
3. ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Включить прибор для прогрева в течение 15 минут. За это время приготовить эталонные растворы.
Приготовление эталонных растворов
В три мерные колбы вместимостью 50 мл отмерить из полумикробюретки 2 мл, 3 мл и 4 мл эталонного раствора дихромата калия, каждый раз, начиная от нуля. Прибавить в каждую колбу по 5 мл серной кислоты (1:1), довести до метки дистиллированной водой, тщательно перемешать. Колориметрировать в кювете с рабочей длиной 3 см, используя светофильтр с длиной волны 340 нм, соответствующей максимальному значению молярного коэффициента поглощения. В качестве раствора сравнения использовать воду.
Измерив оптическую плотность трех эталонных растворов, записать полученные данные в рабочий журнал в таблицу по форме 4. Рассчитать три отношения сэ /Аэ, записать их в таблицу по форме 4 и сравнить между собой.
Форма 4
Результаты измерения оптической плотности эталонных растворов
Содержание хрома в эталонных растворах сэ, мкг Оптическая плотность эталонных растворов Аэ Отношение сэ /Аэ
Учитывая полученный интервал значений величин оптической плотности и предел допустимой погрешности измерения в этой области, равный примерно 1,5-2 %, вычислить относительную погрешность измерения и выбрать результаты для дальнейших расчетов, соответствующие этому критерию. Рассчитать среднее отношение (сэ /Аэ)ср.
112
Определение хрома в исследуемом растворе
Рассчитав среднее отношение концентрации к абсорбционности для эталонных растворов, получить у преподавателя контрольный анализируемый раствор в мерную колбу той же вместимости, добавить к нему то же количество серной кислоты (1:1), довести до метки дистиллированной водой, перемешать и измерить оптическую плотность Ах в тех же условиях, в которых работали с эталонными растворами. Рассчитать содержание хрома в исследуемом растворе сх, используя формулу (1).
Рассчитать абсолютную и относительную D погрешности определения
= m(Сr)ист - m(Сr); D = [ / m(Сr)ист] 100%.
По полученным результатам сформулировать вывод о точности опреде-
ления.
4. CОДЕРЖАНИЕ ОТЧЕТА
Отчет должен содержать:
1.Оптическую схему фотоэлектроколориметра.
2.Экспериментальные данные (таблица по форме 4) и результаты расче-
тов.
2.1.Расчет относительных погрешностей измерения.
2.2.Результаты определения хрома в анализируемом растворе.
3.Расчет абсолютной и относительной погрешностей определения с выводом о точности определения.
Литература: [1], кн. 2, с.50…77.
Вопросы для подготовки к защите лабораторной работы
1.С какой целью в анализируемый раствор вводится кислота?
2.Как зависит величина относительной ошибки определения концентрации от величины пропускания?
3.Как выбирается светофильтр для измерения оптической плотности?
4.При каких условиях целесообразно использование метода сравнения при определении концентрации?
113

ЛАБОРАТОРНАЯ РАБОТА №4 ФОТОКОЛОРИМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ЖЕЛЕЗА МЕТОДОМ КАЛИБРОВОЧНОГО ГРАФИКА
1.ЦЕЛЬ РАБОТЫ
1.Ознакомление с алгоритмом построения фотометрической методики с использованием метода калибровочного графика.
2.Фотоколориметрическое определение содержания железа в виде роданидного комплекса.
2.ОСНОВНЫЕ ТЕОРЕТИЧЕСКИЕ ПОЛОЖЕНИЯ
Метод калибровочного графика используется при многократных серийных определениях. Для получения серии эталонных растворов с возрастающей концентрацией используют метод разбавления исходного первичного эталонного раствора, приготовленного из чистых металлов, солей, стандартных образцов.
Основные этапы работы включают:
 выбор условий фотометрирования − длины волны, соответствующей максимуму светопоглощения и толщины поглощающего слоя;
выбор условий фотометрирования − длины волны, соответствующей максимуму светопоглощения и толщины поглощающего слоя;
 построение калибровочного графика на основе результатов фотометрирования серии эталонных растворов;
построение калибровочного графика на основе результатов фотометрирования серии эталонных растворов;
 определение содержания элемента в исследуемом растворе по построенному графику.
определение содержания элемента в исследуемом растворе по построенному графику.
Рассмотрим алгоритм этого метода на примере определения содержания железа роданидным методом.
Образование роданидного комплекса железа (III) является основой для высокочувствительного метода обнаружения и количественного метода определения этого элемента. Метод позволяет обнаружить даже следовые количества железа, поскольку значение молярного коэффициента поглощения комплекса очень велико.
Образование комплексов железа с роданид-ионами протекает по следующим схемам:
114
Fe3+ + SCN- [FeSCN]2+
[FeSCN]2+ + SCN- [Fe(SCN)2 ]1+
…
[Fe(SCN)5 ]2- + SCN- [Fe(SCN)6 ]3-
При концентрации избытка роданид-ионов с(SCN-) = 5·10-3 моль/л в основном образуется комплекс [FeSCN]2+; при[SCN-] = 1,2·10-2 моль/л − комплекс
[Fe(SCN)2]+, при [SCN-] = 4·10-2моль/л − комплекс [Fe(SCN)3]. Тетра- и пента-
роданиды образуются при концентрации избытка роданид-ионов, равной 0,16 и 0,7 моль/л соответственно. Образующиеся комплексы имеют кроваво-красное окрашивание различной интенсивности.
В связи с невозможностью создать в растворе условия для образования одного комплекса, практически всегда приходится работать с раствором, содержащим одновременно целый ряд железороданидных комплексов, имеющих разные спектральные характеристики. Поэтому во избежание ошибок при определении железа роданидным методом всегда необходимо брать большой избыток реактива и соблюдать одинаковое время для проведения реакции.
Определению мешают вещества, связывающие в комплекс ионы железа или ион роданида (хлориды, фториды, фосфаты, арсенаты, тартраты), восстановители, восстанавливающие железо до железа (II), и окислители, разрушающие роданид-ион. Для предотвращения гидролиза солей железа и возможного при этом выпадения гидроксидов или основных солей железа необходимо подкислять раствор (лучше разбавленной азотной кислотой), так как при большой концентрации нитрат-ионов возможно окисление роданид-иона.
Для измерения оптической плотности растворов используется фотометр с дифракционной решеткой.
3. ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Включить фотометр для прогрева в течение 15 минут. За это время ознакомиться с правилами работы на приборе и приготовить растворы для последующего выполнения первого этапа работы − выбора длины волны, при которой наблюдается максимальное значение оптической плотности.
Выбор длины волны. Максимальное изменение оптической плотности на единицу концентрации в соответствии с законом Бугера-Ламберта-Бера на-
115
блюдается при длине волны, соответствующей максимальному значению коэффициента поглощения. Определить требуемую область длин волн можно из справочной литературы или экспериментально, измерив значения оптической плотности поглощающего раствора при разных длинах волн. Длина волны, при которой получено максимальное значение величины оптической плотности анализируемого раствора, будет использована для проведения всех дальнейших измерений.
Измерение абсорбции эталонного раствора роданида железа необходимо проводить не по отношению к воде (как в работе № 3), а по отношению к смеси всех реактивов, кроме стандартного раствора. Эта необходимость вызвана высоким значением коэффициента поглощения образующегося роданидного комплекса, в результате чего даже следовые количества железа в реактивах могут внести свой вклад в измеряемое значение оптической плотности исследуемого раствора.
Приготовление эталонного раствора. Для приготовления эталонного раствора 1 в мерную колбу вместимостью 100 мл поместить 10 мл исходного эталонного раствора железоаммонийных квасцов с титром по железу, равным Т(Fe) = 0,006968 г/мл. Добавить к раствору 10 мл азотной кислоты с концентрацией 4 моль/л и 10 мл раствора роданида калия с концентрацией с(KSCN) = 6 моль/л. Довести содержимое колбы до метки дистиллированной водой и тщательно перемешать.
Приготовление раствора сравнения. Раствор сравнения следует гото-
вить в такой же мерной колбе, вводя реактивы в той же последовательности, за исключением исходного эталонного раствора железоаммонийных квасцов.
Измерить оптическую плотность эталонного раствора 1 по отношению к раствору сравнения последовательно при длинах волн в пределах от 350 до 650 нм, используя кюветы с рабочей длиной 10 мм. Полученные результаты измерений занести в таблицу, составленную по форме 5.
116

Форма 5
Зависимость величины оптической плотности от длины волны излучения
Длина волны излучения λ, нм Значения оптической плотности А
На основе анализа полученных данных следует построить спектр поглощения в координатах А - λ, выбрать длину волны для измерения абсорбции и приступить к построению калибровочного графика.
Построение калибровочного графика
В три пронумерованные мерные колбы вместимостью 100 мл отмерить 10, 20 и 30 мл исходного стандартного раствора железоаммонийных квасцов. Порядок приготовления эталонных растворов тот же, что и при выборе светофильтра. Раствор сравнения и кюветы те же.
Примечание
1.Фотометрирование необходимо проводить сразу же после приготовления раствора, поскольку комплекс быстро разрушается. Поэтому растворы готовятся последовательно, только после окончания фотометрирования предыдущего.
2.Каждый раствор готовится и фотометрируется трижды.
3.Мерные колбы необходимо очень тщательно мыть перед приготовлением каждого последующего раствора. Кюветы с анализируемым раствором перед заполнением несколько раз ополаскивать этим раствором.
4.Кювета с раствором сравнения используется одна и та же на протяжении всей работы. Измерение оптической плотности производится при длине волны, выбранной в первой части работы.
Результаты фотометрирования по мере измерения заносятся в таблицу по форме 6.
117

Форма 6
Оптическая плотность эталонных растворов
Объем исходного эталонного раствора Vэт, мл Значения величин оптической плотности Аэт.1
Аэт.2
Аэт.3
Аэт. Ср
По полученным данным построить калибровочный график в координатах Аэт/Vэт. Масштаб графика должен соответствовать точности измерений.
Определение содержания железа в анализируемом растворе
В мерную колбу вместимостью 100 мл получить у преподавателя исследуемый раствор, добавить в него 10 мл азотной кислоты с концентрацией с(HNO3) = 4 моль/л, 10 мл раствора роданида калия, довести до метки дистиллированной водой, тщательно перемешать и измерить оптическую плотность раствора, используя тот же раствор сравнения и те же кюветы, что и при построении калибровочного графика.
Расчет содержания железа
Содержание железа в анализируемом растворе определяется по формуле: m(Fe) = T(Fe) · V ,
где m(Fe) − содержание железа в анализируемом растворе; T(Fe) − титр исходного стандартного раствора железоаммонийных квасцов; V − объем стандартного раствора, найденный по калибровочному графику в соответствии с измеренной величиной оптической плотности Ах.
4. СОДЕРЖАНИЕ ОТЧЕТА
Отчет должен содержать:
1.Оптическую схему фотоэлектроколориметра.
2.Данные измерений оптической плотности эталонного раствора для выбора длины волны, соответствующей максимуму поглощения (форма 5).
118
3.Данные измерений оптической плотности серии эталонных растворов для построения калибровочного графика (форма 6) и калибровочный график зависимости оптической плотности от объема эталонного раствора.
4.Результаты измерения оптической плотности анализируемого раствора
ирасчет содержания железа в этом растворе.
Литература: [1], кн. 2, с. 50…76
Вопросы для подготовки к защите лабораторной работы
1.Как могут влиять на линейность калибровочного графика время, предшествующее измерению, изменение величины рН раствора?
2.Какие моменты в методике необходимо строго соблюдать, чтобы получить линейный калибровочный график?
3.Допустимо ли получение точки на калибровочном графике по результатам одного измерения?
4.Как выбирается светофильтр при фотометрических определениях?
5.Когда целесообразно использовать метод калибровочного графика для анализа?
6.Перечислите основные этапы построения аналитической методики при использовании метода калибровочного графика для определения концентрации анализируемого вещества.
119
ЛАБОРАТОРНАЯ РАБОТА №5 ФОТОКОЛОРИМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ НИКЕЛЯ В СТАЛЯХ
1.ЦЕЛЬ РАБОТЫ
1.Освоение принципов построения методики элементного анализа сталей фотометрическим методом.
2.Определение содержания никеля в сталях методом калибровочного графика.
2.ОСНОВНЫЕ ТЕОРЕТИЧЕСКИЕ ПОЛОЖЕНИЯ
Трудности анализа реальных веществ в фотометрии обусловлены сложностью и разнообразием их состава. Введение каждого нового компонента вносит в задачу сразу несколько новых переменных. При выборе метода анализа первый этап состоит в четком выяснении и формулировании аналитической задачи, что в основном будет определяться ответами на следующие вопросы.
1.Каков интервал концентраций определяемых компонентов?
2.Какова требуемая степень точности результатов анализа?
3.Какие компоненты из сопутствующих определяемому обладают сходными химико-аналитическими свойствами?
4.Какие химические и физические свойства анализируемого материала должны быть учтены при выборе метода анализа?
5.Сколько проб и за какой срок должно быть проанализировано?
Только одновременное рассмотрение и учет названных факторов с позиций системного анализа может обеспечить обоснованный выбор метода анализа. Следовательно, для выбора метода количественного определения элемента в природном или техническом материале необходимо предварительно располагать информацией о его качественном и полуколичественном составе. Затем составляется полная схема анализа исследуемого объекта, которая включает методы разложения пробы, удаление или маскирование мешающих компонентов, перевод определяемого элемента в удобную для анализа форму.
Анализ проводится по рабочей прописи − методике, которая представляет собой подробное описание всех операций и условий проведения определения, выполнение которых должно обеспечить регламентированные характеристики результатов анализа.
120
Методика анализа
Методика анализа включает:
1.Отбор средней пробы.
2.Взятие навески или измерение объема раствора для жидкой пробы.
3.Растворение пробы (в воде, в минеральных кислотах или их смесях, в щелочи) или разложение сплавлением (кислым или щелочным) с последующим переведением в раствор.
4.Отделение мешающих компонентов или их маскирование.
5.Проведение аналитической реакции.
6.Измерение аналитического сигнала.
7.Расчет содержания определяемого элемента и необходимых метрологических характеристик.
Методы определения никеля в черных металлах и сплавах
Для определения никеля в черных металлах и сплавах в зависимости от ожидаемого количественного содержания элемента рекомендуются следующие методы определения:
1.Фотометрический (при содержании никеля от 0,05 до 3 %), на основе реакции с диметилглиоксимом;
2.При содержании никеля от 1 до 30 % может быть использован гравиметрический метод, основанный на осаждении никеля диметилглиоксимом в аммиачной среде.
3.Комплексонометрический, основанный на титровании никеля комплексоном III в аммиачной среде в присутствии индикатора мурексида.
Фотометрический метод основан на измерении интенсивности окраски комплексного соединения никеля с диметилглиоксимом в щелочной среде (рН = 7-12) в присутствии окислителя − надсернокислого аммония. Окислитель повышает чувствительность реакции, однако большой избыток окислителя разрушает полученное соединение. Окислитель и щелочь необходимо прибавлять последними к уже готовой смеси в связи с тем, что образующееся соединение очень неустойчиво.
Определению мешают окрашенные вещества и катионы, образующие нерастворимые гидроксиды. Для отделения от мешающих компонентов никель экстрагируют хлороформом в присутствии лимонно-кислого натрия и гидроксиламина в виде диметилглиоксимата. Влияние меди и кобальта устраняют
121
промыванием хлороформного раствора аммиаком. Никель реэкстрагируют хлороводородной кислотой.
В отсутствие кобальта влияние остальных мешающих компонентов, и в первую очередь железа, устраняют путем переведения их в вино-кислые комплексы.
3. ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ Приготовление анализируемого раствора
На аналитических весах берут навеску стали массой 0,1 г с точностью до четвертой значащей цифры. Навеску помещают в коническую колбу вместимостью 100 − 150 мл, прибавляют примерно 10 −15 мл хлороводородной кислоты с концентрацией (1:1) и нагревают на песчаной бане до прекращения реакции. Добавляют несколько капель концентрированной азотной кислоты для окисления железа и нерастворившихся карбидов. Раствор кипятят в течение 3 − 5 минут, осторожно обмывают стенки колбы водой и выпаривают почти до конца до получения влажного осадка.
Остужают колбу в вытяжном шкафу, обмывают стенки водой и количественно переносят содержимое в мерную колбу вместимостью 100 мл. Разбавляют до метки дистиллированной водой, перемешивают и отбирают пипеткой две аликвотные части по 5-10 мл в мерные колбы вместимостью 100мл. В каждую колбу приливают при перемешивании 10 мл раствора сегнетовой соли с содержанием 200 г/л и 10 мл раствора едкого натра (50 г/л). Тщательно перемешав содержимое колб, добавляют в каждую по 10 мл надсернокислого аммония (30 г/л) и по 10 мл щелочного раствора диметилглиоксима (10 г/л). Спустя три минуты содержимое колб разбавляют дистиллированной водой до метки, тщательно перемешивают и через 5-7 минут измеряют оптическую плотность раствора.
Фотометрирование раствора производится при длине волны 430...450
нм.
В качестве раствора сравнения используют аликвотную часть раствора анализируемой пробы, в которую добавлены все реактивы, за исключением щелочного раствора диметилглиоксима, вместо которого добавлено 10 мл едкого натра.
По готовому калибровочному графику определяют содержание никеля в анализируемом растворе с(Ni), соответствующее найденному значению оптической плотности Ах.
122
Расчет содержания никеля
Расчет содержания никеля в анализируемом образце производят по фор-
муле:
(Ni) = с(Ni) 10-6 · Vк · 100 / mстали · Vп,
где mстали навеска стал,и г; с(Ni), − содержание никеля, мкг, в анализируемом растворе; Vк − вместимость мерной колбы, мл; Vп − вместимость пипетки (объ-
ем аликвотной части, мл); (Ni) − массовая доля никеля, %.
Расчет абсолютной и относительной погрешности определения и вывод, характеризующий результаты работы
Расчет абсолютной и относительной D погрешности определения производится по формулам
= (Ni) ист - (Ni);
D = [ / (Ni)ист] 100% .
4.СОДЕРЖАНИЕ ОТЧЕТА
Отчет должен содержать:
1.Оптическую схему фотоэлектроколориметра.
2.Краткую методику определения.
3.Данные измерения оптической плотности.
4.Расчет содержания никеля в исследуемом образце стали.
5.Расчет абсолютной и относительной погрешности определения и вывод, характеризующий результаты работы.
Литература: [1], кн. 2 , с. 50 …76.
Вопросы для подготовки к защите лабораторной работы
1.Перечислить основные этапы построения аналитической методики при фотометрических определениях в сплавах и рудах.
2.Какие факторы учитываются при подборе эталонов для построения калибровочного графика?
3.В каких случаях для построения калибровочного графика используются стандартные образцы, чистые металлы, соли?
4.Объясните роль каждого реагента, используемого в ходе определения.
123
1.2.2. Атомно-эмиссионный спектральный анализ (лабораторные работы № 6…8)
Методы атомно-эмиссионной спектроскопии основаны на изучении спектров испускания атомов, возникающих при испарении пробы.
В основном состоянии атомы, молекулы и ионы всех элементов обладают минимальной энергией Ео и не испускают излучения. Возбуждение, происходящее под влиянием внешних воздействий (столкновение с другими частицами, ионами, электронами, при облучении электромагнитным излучением), сопровождается переходом внешних, валентных электронов с основного на один из более высоких энергетических уровней. Но в возбужденном состоянии электроны могут находиться в течение очень короткого времени. По истечении примерно 10-8 с возбужденный атом возвращается в основное или в какое-либо промежуточное энергетическое состояние. Энергия, высвобождающаяся при этом, испускается в виде кванта и может быть зафиксирована в виде отдельной спектральной линии.
Излучение длины волны, соответствующее определенному энергетическому переходу, называется спектральной линией.
Для возбуждения спектральной линии необходима определенная энергия, называемая потенциалом возбуждения.
Энергия, необходимая для полного отрыва электрона от атома и перевода его в ионное состояние, называется потенциалом ионизации.
Эмиссионные спектры каждого элемента представляют собой набор спектральных линий с различными длинами волн, соответствующих различным энергетическим переходам.
Длины волн такого спектра характерны только для данного элемента, они носят сугубо индивидуальный характер. Число всех возможных электронных переходов, а следовательно, и число линий в спектре данного элемента определяется числом и размещением внешних электронов. Атомы с малым числом внешних электронов (например, натрий или литий) имеют простой спектр, состоящий всего из нескольких линий. Атомы со сложно построенными внешними оболочками − особенно элементы побочных подгрупп периодической системы − дают сложные спектры с очень большим числом линий. Так, в спектрах d- и f-элементов, например железа, насчитываются десятки тысяч спектральных линий, которые отчетливо воспроизводятся.
124
Линии излучения, соответствующие атомам различных элементов, сведены в таблицы (атласы) спектральных линий.
В общеупотребительных таблицах спектральных линий, «Спектральных атласах», приводятся от 50000 до 100000 линий.
Атомно-эмиссионный спектральный анализ используется для проведения качественного и количественного элементного анализа.
Качественный спектральный анализ основан на том, что атомы каждого элемента характеризуются вполне определенным набором спектральных линий, данные по которым сведены в специальные таблицы и атласы.
Количественный спектральный анализ основан на существовании зависимости интенсивности каждой спектральной линии от концентрации атомов в плазме разряда.
Принципиальная схема аппаратуры
Принципиальная схема аппаратуры атомного эмиссионного спектрального анализа включает:
 Источник возбуждения спектра
Источник возбуждения спектра
Традиционными, хорошо методически отработанными способами атомизации анализируемого образца являются пламя, дуга и искра. В последние годы в связи с появлением новых задач появился целый ряд новых источников, таких как источник индуктивно-связанной плазмы (ИСП), микроволновый разряд, лазерные атомизаторы.
 Диспергирующее устройство
Диспергирующее устройство
Для диспергирования излучения используют призмы и дифракционные решетки.
 Регистрирующее устройство
Регистрирующее устройство
Регистрация спектра может проводиться визуальным, фотографическим и фотоэлектрическим способами.
Визуальный вариант регистрации спектра обеспечивает получение качественной и полуколичественной информации. Приборы, используемые для этой цели, называются стилоскопами.
Наиболее полную информацию качественного и количественного характера можно получить при фотографической регистрации спектра. Используемые
125
для этой цели приборы называются спектрографами. Однако этот способ проведения спектрального анализа требует слишком много времени и потому в современной аналитической службе металлургического и машиностроительного предприятия при серийных анализах однородных материалов основным, безус-
ловно, является эмиссионный спектральный анализ с фотоэлектрической регистрацией спектра. Аппаратура этого класса называется полихроматорами или
квантометрами.
Качественный анализ можно проводить фотографическим или визуальным способом. В первом случае эмиссионные спектры фотографируются на фотопластинку или фотопленку. Данные, полученные при этом, являются объективными и документальными, могут храниться любое время и при необходимости могут быть перепроверены. Качественный анализ проводится сравнением спектра исследуемого образца со спектром известных элементов или со спектром железа, которые фотографируются встык со спектром пробы (рис. 1). При визуальном способе наблюдают спектр исследуемого образца непосредственно в окуляре стилоскопа.
Для проведения качественного анализа необходимо установить наличие или отсутствие в спектре так называемых аналитических или последних линий. При уменьшении концентрации элемента в пробе эти линии исчезают последними. Они хорошо изучены, их длины волн и характеристику интенсивности можно найти в специальных справочниках. Чаще всего это резонансные линии, соответствующие переходу из первого возбужденного состояния в основное, то есть обладающее наименьшей энергией.
126

Рис. 1. Спектр железа и спектр марганца
127

Обязательным условием для проведения и качественного, и количественного анализа является использование таких аналитических линий, которые не накладываются на линии других элементов. Если по этой причине невозможно использовать последние линии, то необходимо использовать другие, менее чувствительные, но не накладывающиеся линии.
Количественный спектральный анализ основан на том, что интенсив-
ность спектральной линии I зависит от концентрации элемента с. Связь между этими величинами нелинейна и выражается в виде уравнения Ломакина:
I = а св ,
где а и в − эмпирические коэффициенты.
Следует заметить, что интенсивность спектральных линий зависит не только от содержания элемента в пробе, но и от условий возбуждения спектра. Поэтому количественные определения проводят по относительной интенсивности линии. Под этим понимают отношение интенсивности линии определяемого элемента к интенсивности другой спектральной линии, называемой линией сравнения. Линия сравнения должна принадлежать спектру элемента, содержание которого в пробе не меняется. Обычно это либо элемент основы, либо специально введенный внутренний стандарт. Линии определяемого элемента и элемента сравнения называют аналитической парой.
Успех количественного спектрального анализа в значительной степени зависит от правильного выбора этой пары линий. Обе линии должны быть гомологичны и удовлетворять следующим правилам:
 расположены в спектре недалеко друг от друга;
расположены в спектре недалеко друг от друга;
 не должны сильно различаться по абсолютной интенсивности;
не должны сильно различаться по абсолютной интенсивности;
 иметь близкие значения потенциалов ионизации.
иметь близкие значения потенциалов ионизации.
Относительная интенсивность аналитической пары линий зависит только от концентрации определяемого элемента в образце.
Низкий предел обнаружения позволяет определять примеси в веществах высокой чистоты (при содержании некоторых элементов 10-5 – 10-7 %), например анализировать чистые и особо чистые вещества в металлургии при производстве атомных и полупроводниковых материалов. Высокая селективность анализа позволяет на одних и тех же приборах определять различные элементы в одной и той же пробе, выбирая в каждом отдельном случае наиболее благо-
128
приятные условия для получения оптимальных метрологических характеристик метода.
Однако метод эмиссионного спектрального анализа не может быть использован для определения высоких содержаний элементов в пробе. Так, при определении содержания алюминия в полученном электролизом металле эмиссионный метод дает неверные результаты (8-12 % вместо реальных 99-99,9 %). Подобные результаты характерны и для других сплавов.
Для определения высоких концентраций элементов в сплавах используются, например, химические, фотометрические или рентгенофлуоресцентные методы.
129
ЛАБОРАТОРНАЯ РАБОТА №6 ПЛАМЕННО-ЭМИССИОННАЯ СПЕКТРОМЕТРИЯ. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ЩЕЛОЧНЫХ МЕТАЛЛОВ
1.ЦЕЛЬ РАБОТЫ
1.Ознакомление с алгоритмом построения аналитической методики при использовании метода пламенно-эмиссионной спектрометрии в определении щелочных металлов.
2.Пламенно-эмиссионное определение содержания щелочных металлов.
2.ОСНОВНЫЕ ТЕОРЕТИЧЕСКИЕ ПОЛОЖЕНИЯ
Метод пламенно-эмиссионной спектрометрии (пламенной фотометрии) является практически единственным методом для определения содержания щелочных металлов в различных природных и технических объектах.
Пламенная фотометрия основана на возбуждении в пламени спектра определяемого элемента и непосредственном измерении интенсивности свечения аналитической линии, пропорциональной содержанию элемента в анализируемом растворе в определенном интервале концентраций.
В зависимости от состава горючей смеси пламя обеспечивает температуру в интервале 2000 − 3000 К. Эта температура значительно ниже, чем в дуге или искре. Поэтому в пламени возбуждаются только наиболее чувствительные спектральные линии с низкими потенциалами возбуждения (не более 5 эВ). Изменяя условия горения, этим методом можно определить более 70 элементов. Однако интенсивность линий большинства элементов, а соответственно и чувствительность регистрации мала. Поэтому на практике число элементов, определяемых этим методом, значительно меньше, чем при возбуждении электрическими источниками света. В основном метод используется для определения щелочных металлов, для которых практически нет экспрессных химических методов анализа, а также для щелочно-земельных элементов.
Эмиссионные спектры этих элементов, получаемые в пламени состоят из нескольких спектральных линий с характерной для каждого элемента длиной волны. Это позволяет использовать метод и для качественного, и для количественного анализа.
Аппаратура. В пламенном фотометре любого типа имеется три основных системы: возбуждения, выделения аналитической спектральной линии и регистрации интенсивности излучения линии.
130
Система возбуждения. Система возбуждения спектральных линий включает распылитель и распылительную камеру, смеситель − отстойник, горелку и пламя. Анализируемый раствор вводится в пламя в виде аэрозоля.
Когда аэрозоль попадает в пламя, протекает ряд быстро сменяющих друг друга процессов:
 испарение растворителя с образованием твердых частиц сухого вещества (солей);
испарение растворителя с образованием твердых частиц сухого вещества (солей);
 разложение или испарение твердых частиц с диссоциацией части молекул с образованием нейтральных атомов, а в некоторых случаях и ионов;
разложение или испарение твердых частиц с диссоциацией части молекул с образованием нейтральных атомов, а в некоторых случаях и ионов;  соединение некоторых атомов с другими атомами или радикалами,
соединение некоторых атомов с другими атомами или радикалами,
присутствующими в пламени;
 возбуждение атомов и некоторых молекул за счет тепловой энергии пламени;
возбуждение атомов и некоторых молекул за счет тепловой энергии пламени;
 возвращение атомов в исходное состояние с выделением квантов
возвращение атомов в исходное состояние с выделением квантов
света.
Для возбуждения атомов пламя должно иметь достаточно высокую температуру и давать достаточное количество тепла. В противном случае за счет расходования энергии на стадиях, предшествующих возбуждению, частицы быстро охлаждаются. Необходимая для возбуждения атомов щелочных металлов температура 1700 - 1900 °С достигается при сжигании смесей воздуха с пропаном и бутаном. Для определения щелочно-земельных элементов необходимо использовать смесь ацетилен − воздух, дающую температуру около
2300 °С.
Система выделения спектральной линии. Система выделения спек-
тральной линии состоит из светофильтров или спектральных приборов − монохроматоров. Максимум пропускания светофильтра должен совпадать с длиной волны спектральной линии или молекулярной полосы определяемого элемента. При необходимости разделения нескольких близко расположенных спектральных линий применяют монохроматоры − спектральные приборы, у которых на выходе установлены щели, которые позволяют выделить необходимые линии (пламенные спектрофотометры).
Система регистрации интенсивности излучения спектральной ли-
нии. Система регистрации интенсивности излучения спектральной линии включает фотоэлементы или фотоэлектроумножители, усилительные и регистрирующие приборы.
131

Интенсивность излучения атомами (молекулами) пропорциональна их концентрации в пламени, которая в свою очередь пропорциональна концентрации ионов в растворе.
Этот метод используется для определения содержания щелочных и ще- лочно-земельных элементов при анализе сырья и продукции металлургической промышленности. Например, в технологической схеме производства алюминия пламенная фотометрия используется для определения содержания натрия.
Необходимая для возбуждения атомов этих металлов температура 1700 – 1900 °С достигается при сжигании смесей воздуха с пропаном и бутаном. Для определения щелочно-земельных элементов необходимо использовать смесь ацетилен-воздух, дающую температуру около 2300 °С.
Для определения можно использовать как пламенные фотометры со светофильтрами, так и спектрофотометры для пламенной фотометрии. Из спектра эмиссии монохроматором (в фотометрах светофильтрами) выделяются характерные для определяемых элементов линии: для определения натрия
= 589 нм, калия = 768 нм, лития =671 нм, кальция = 622 нм, стронция
= 460 нм, магния = 384 нм.
Для щелочных металлов чувствительность перед обнаружением достигает 0,1-0,001 мкг/мл. Точность определений 2 – 4 % .
Определение содержания проводят методом калибровочного графика или методом добавок.
Краткое описание прибора4
Для анализа могут быть использованы разные марки отечественных и зарубежных фотометров и спектрофотометров для пламенной фотометрии. Описание прибора и инструкция по работе на конкретном приборе находятся на рабочем месте.
4 Включение прибора, подготовку его к работе, подключение баллона с газом и компрессора для подачи сжатого воздуха должен выполнять только преподаватель или инженер, обслуживающий данную установку.
132
3. ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Подготовка прибора
Основные действия:
1.Подключение к компрессору, подающему сжатый воздух, и к газовому баллону.
2.Включение анализатора в сеть.
3.Зажигание и регулировка пламени таким образом, чтобы внутренний конус был голубовато-зеленым, пламя горело спокойно, без мерцания.
4.Установка нужного светофильтра.
5.Установка нулевого деления на шкале при впрыскивании в пламя дистиллированной воды.
6.Установка требуемой чувствительности прибора, при которой максимальное отклонение стрелки амперметра соответствует максимально возможному содержанию элемента в анализируемой пробе, то есть максимальной концентрации стандартного раствора.
Построение градуировочного графика
Концентрация определяемых металлов в приготовленных первичных стандартных растворах должна быть 1000 мг/ л.
Рабочие стандартные растворы, используемые для построения градуировочного графика, готовят разбавлением первичных стандартных растворов в мерных колбах вместимостью 100 мл, беря для этого 10, 15, 25, 50 мл исходного стандартного раствора.
Приготовленные рабочие стандартные растворы помещают в чистые полиэтиленовые стаканчики, ополоснутые этими же растворами.
Промыв прибор дистиллированной водой, поочередно измеряют интенсивность излучения рабочих стандартных растворов, начиная с самого разбавленного. Каждое измерение повторяют 2-3 раза. Результаты измерений сразу заносят в таблицу по форме 7. По полученным данным строят калибровочный график, откладывая по оси абсцисс значения концентрации определяемого элемента в мкг/мл, а по оси ординат - показания микроамперметра.
Определение содержания заданного элемента в анализируемом растворе
Приготовленный анализируемый раствор вводят в пламя после предварительного промывания прибора и измеряют показания микроамперметра. Изме-
133
рение повторяют 2-3 раза и по градуировочному графику определяют соответствующее значение концентрации в измеренном растворе.
Форма 7
Результаты измерения интенсивности излучения рабочих стандартных растворов
Концентрации стандартных растворов, мкг/мл Показания микроамперметра, мкА
4. СОДЕРЖАНИЕ ОТЧЕТА
Отчет должен содержать:
1.Принципиальную схему прибора.
2.Данные измерений интенсивности излучения рабочих стандартных растворов (форма 7) и калибровочный график.
3.Результаты измерения интенсивности излучения анализируемого раствора и расчет содержания определяемого элемента в анализируемом объекте.
Литература: [1], кн. 2, с.48..50.
Вопросы для подготовки к защите лабораторной работы
1.Перечислите основные атомно-эмиссионные и атомно-абсорбционные методы анализа.
2.Охарактеризуйте основные процессы, протекающие в пламени.
3.Дайте характеристику основных типов помех, возникающих в процессе определения пламенно-эмиссионным методом.
4.Почему метод пламенной фотометрии используется только для определения щелочных и щелочно-земельных элементов?
134
ЛАБОРАТОРНАЯ РАБОТА №7 КАЧЕСТВЕННЫЙ ЭМИССИОННЫЙ СПЕКТРАЛЬНЫЙ АНАЛИЗ.
ПОЛУКОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ЦИНКА В ЛАТУНИ И БРОНЗЕ, ХРОМА В ЧУГУНЕ И СТАЛИ С ПОМОЩЬЮ СТИЛОСКОПА
1.ЦЕЛЬ РАБОТЫ
1.Ознакомление с методом качественного и полуколичественного визуального спектрального анализа.
2.Полуколичественное определение цинка в латуни и бронзе, хрома в чугуне и стали.
2.ОСНОВНЫЕ ТЕОРЕТИЧЕСКИЕ ПОЛОЖЕНИЯ
Стилоскоп − это спектроскоп для проведения качественного и полуколичественного визуального спектрального анализа. В первую очередь, этот прибор был предназначен для определения сортов образцов стали, что и определило его название “steelscope” (steel − по английски сталь). Разработанный и внедренный в практику еще в начале 1930 - х гг. прибор и до сих пор широко применяется в практике в производственных условиях, прежде всего, для сортировки сплавов. При этом не требуется отбирать пробу в виде стружки. Можно анализировать готовую деталь, слиток, заготовку и пр. Большую пользу применение стилоскопов приносит при контроле сплавов, поступающих на предприятие. При этом осуществляется либо сплошной контроль металла, либо проверка внушающих подозрение партий сплавов. Стилоскопы применяют на складах, шихтовых дворах, в пунктах сортировки металлического лома. Рационально его применение в цехах металлообрабатывающих заводов для организации сплошного и выборочного контроля материалов в разнообразных звеньях технологического процесса, проведения анализа заготовок, полуфабрикатов, готовых деталей.
Как уже было сказано, в основе эмиссионного спектрального анализа лежат два положения:
 атомы каждого элемента характеризуются определенным набором спектральных линий;
атомы каждого элемента характеризуются определенным набором спектральных линий;
 интенсивность каждой спектральной линии зависит от концентрации атомов в плазме разряда.
интенсивность каждой спектральной линии зависит от концентрации атомов в плазме разряда.
Однако многие спектральные линии различных элементов располагаются очень близко и при использовании приборов со средней дисперсией различить их практически невозможно. В этом случае говорят о наложении линий, ме-
135
шающем их правильному отождествлению. Это характерно для анализа сложнолегированных сталей, жаропрочных сплавов, сложных руд с большим содержанием железа. Уменьшить эффект наложения позволяет использование приборов с большой дисперсией.
При проведении качественного анализа можно использовать любые подходящие линии, исключая эффект наложения.
Однако для определения малых (следовых) содержаний элементов необходимо пользоваться наиболее интенсивными последними линиями. Списки «последних» линий приводятся в справочниках и таблицах.
Принцип стилоскопического полуколичественного анализа заключается в визуальном сравнении яркостей линий анализируемой примеси и одной или нескольких линий элемента основы. При этом линии аналитической пары должны располагаться близко друг к другу и не должны сильно различаться по цвету. Приблизительное равенство яркостей сравниваемых линий позволяет определить некоторый интервал концентраций определяемого элемента.
В современных моделях стилоскопов для облегчения работы и увеличения точности определения эти линии могут выноситься на экран дисплея.
При первоначальном рассматривании спектра с большим числом линий, например спектра железа, создается впечатление, что разобраться в таком количестве линий просто невозможно. Однако последующее наблюдение показывает, что отдельные спектральные линии имеют свои индивидуальные особенности: различный внешний вид − отчетливость, контрастность, размытость; различную интенсивность.
Для анализа спектр необходимо, прежде всего, расчленить на отдельные характерные группировки, а затем уже проводить анализ внутри них по отдельным спектральным линиям.
Конечно, визуальный спектральный анализ имеет свои ограничения. Совершенно нецелесообразно использовать его для анализа сложных незнакомых спектров, при минимальной возможности повреждения образца или при очень малом количестве пробы. Во всех подобных случаях следует сфотографировать спектр.
Оптическая схема стилоскопа
Стилоскоп СЛ-11 имеет автоколлимационную оптическую схему. Излучение от источника (дуги или искры) направляется на щель постоянной ширины (0,02 мм), прорезанной на металлическом слое, нанесенном на стеклянную пластинку. Диспергирующая призма закреплена неподвижно, а вторая, боль-
136

шой катет которой покрыт зеркальным слоем, может поворачиваться, вследствие чего спектр перемещается в поле зрения окуляра. В фокальной плоскости окуляра расположен фотометрический клин. Спектр наблюдается через окуляр. Фотометрический клин виден в поле зрения окуляра в виде узкой полоски.
Для определения значения длин волн пользуются предварительно построенной дисперсионной кривой стилоскопа, которая представляет собой зависимость между длиной волны излучения (линий спектра) и делением шкалы барабана. Обычно дисперсионную кривую строят по спектру железа. В деталях такие кривые индивидуальны для каждого прибора.
Качественный анализ на стилоскопе
Сортировка латуни и бронзы
Латунь представляет собою сплав с содержанием цинка до 35…40 %, а в безоловянистой бронзе его содержание не превышает 5 %. Внешне образцы практически неотличимы, но результаты стилоскопического анализа по оценке содержания цинка в анализируемых образцах могут служить основой для заключения о составе сплава.
Химический состав эталонных образцов латуни, содержащей цинк, и бронзы, содержащей только следы цинка, представлен в табл. 1.
|
|
|
|
|
|
|
Таблица 1 |
|
Химический состав цветных сплавов на основе меди |
|
|||||
|
|
|
Химический состав образцов, % |
|
|||
Образец |
|
|
|
||||
|
|
|
|
|
|
|
|
|
Zn |
Si |
Al |
Mn |
|
Cu |
|
|
|
|
|||||
Латунь |
|
16 |
4 |
|
|
|
80 |
|
|
|
|
|
2 |
|
|
Бронза |
|
0.2 |
|
9 |
|
|
|
При стилоскопическом определении цинка изучают красную область спектра. Присутствие цинка в сплавах устанавливается по появлению красной линии цинка 636,2 нмв группе Zn1. Индекс «1» (Zn1) означает, что при возбуждении спектра в дуге эта линия исчезает последней, индекс «2» указывает на то, что данная линия исчезает предпоследней и т.д. В латунях, где содержание цинка составляет более 10 %, эта линия обладает столь большой яркостью, что возникает впечатление, словно в этой области отсутствуют какие-либо иные спектральные линии. Только при очень внимательном рассмотрении спектра удается заметить несколько линий меди. Для этой же цели может быть использована группа Zn2. Группа Zn2 позволяет оценить содержание цинка в широком диапазоне концентраций.
137

Однако если отсортировываются оловянистые бронзы, где содержание цинка может доходить до 14 %, то отличить их по красной линии 636,23 нм от латуней с содержанием цинка около 40 % можно только при очень большом навыке.
Более надежно сделать это по группе Zn2 (синяя область спектра) и особенно по группе Zn3.
Полуколичественный анализ на стилоскопе. Полуколичественное определение содержания хрома в чугуне и стали
В основе полуколичественного стилоскопического анализа лежит сравнение яркостей линий аналитической пары, то есть линии анализируемой примеси с одной или несколькими линиями основы. Точное равенство яркостей линий оценить визуально, конечно, невозможно. Поэтому определяется некоторый интервал концентраций определяемого элемента.
Так, при определении хрома в сталях сравнивают интенсивности линий хрома и железа (элемента основы). Если содержание хрома меняется в широких пределах, то для его оценки используют фиолетовую, синюю и зеленую области спектра (табл. 2).
Таблица 2
Некоторые стилоскопические признаки для оценки содержания хрома в сталях
Содержание |
Длина волны, нм, |
Содержание |
Длина волны, нм, |
|
хрома в стали, % |
хрома железа |
хрома в стали, % |
хрома железа |
|
|
|
|
|
|
0,1 |
520,84 = 520,23 |
1,5 |
– 2,2 |
465,22 ≥ 465,45 |
|
|
|
|
|
0,2 |
520,60 = 520,23 |
|
|
540,98 = 541,52 |
|
|
|
|
|
0,1 – 0,3 |
425,43 = 424,74 |
2,5 |
– 8,0 |
540,98 = 540,58 |
|
|
|
|
|
0,3 – 0,7 |
464,62 < 464,74 |
|
|
435,10 < 437,59 |
|
|
|
|
|
|
465,22 ≤ 464,35 |
8,0 – 13,0 |
435,10 = 437,59 |
|
|
|
|
|
|
0,7 – 1,1 |
464,62 = 464,74 |
|
|
435,10 ≤ 435,27 |
|
|
|
|
|
|
465,22 < 465,45 |
13,0 |
– 20,0 |
435,10 = 435,27 |
|
|
|
|
|
|
540,98 < 541,09 |
|
|
492,23 < 491,90 |
|
|
|
|
|
1,0 – 1,6 |
465,22 ≤ 465,45 |
> 20,0 |
492,232 ≥ 491,90 |
|
|
|
|
|
|
|
540,98 = 541,09 |
|
|
|
Оценка яркостей первой и второй линий в таблице проведена знаками: =, <, >.
138

3.ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Взависимости от полученного задания: качественный или полуколичественный анализ, − выполнить вариант 1 или вариант 2.
Вариант 1
1.Получить у преподавателя образцы, подлежащие сортировке.
2.Выбрать линии спектра, по которым будет производиться сортировка. По дисперсионной кривой определить, какие деления шкалы барабана соответствуют определяемым линиям спектра.
3.Установить электроды, предварительно зачистив их шкуркой. В качестве электрода сравнения устанавливается медный диск, края которого предварительно зачищаются. На анализируемых деталях необходимо выбрать небольшой плоский участок и хорошо зачистить его.
4.Установить на шкале барабана требуемое деление, включить дугу и рассмотреть в окуляр требуемый участок спектра. Оценить присутствие или отсутствие цинка в анализируемом образце.
5.Последовательно рассмотреть спектры всех анализируемых образцов.
6.Результаты анализа занести в таблицу по форме 8 и сформулировать вывод по результатам изучения спектров.
|
|
|
Форма 8 |
Стилоскопическая сортировка образцов медных сплавов |
|||
|
|
Деление шкалы |
|
Искомый элемент |
Выбранная линия |
Оценка |
|
|
спектра, нм |
барабана |
присутствия эле- |
|
|
|
|
|
|
|
|
|
|
|
|
Вариант 2 1. В качестве электрода сравнения устанавливается диск из чистого желе-
за.
2.Последовательно рассмотреть спектр анализируемого образца в соответствии с интервалами длин волн, указанными в табл. 2.
3.Результаты анализа занести в таблицу по форме 9 и сформулировать вывод по результатам изучения спектров.
139

Форма 9
Стилоскопическая оценка содержания хрома в анализируемом образце стали
Предполагаемое |
Выбранные линии |
Деления шкалы |
Оценка содержа- |
содержание хрома |
спектра, нм, |
барабана |
ния хрома |
|
Cr-Fe |
Cr-Fe |
|
|
|
|
|
|
|
|
|
4. СОДЕРЖАНИЕ ОТЧЕТА
Отчет должен содержать:
1.Цель работы.
2.Оптическую схему стилоскопа.
3.Данные анализа по форме 8 или 9.
Литература: [1], кн. 2, с. 31…32, 40…41.
Вопросы для подготовки к защите лабораторной работы
1.Перечислите основные атомно-эмиссионные и атомно-абсорбционные методы анализа.
2.Что представляет собой дисперсионная кривая стилоскопа ?
3.На чем основан качественный атомно-эмиссионный анализ?
4.Какие линии называются последними?
5.Какую информацию можно получить при стилоскопическом анализе?
140
ЛАБОРАТОРНАЯ РАБОТА №8 АТОМНО-ЭМИССИОННЫЙ СПЕКТРАЛЬНЫЙ АНАЛИЗ ЛЕГИРОВАННОЙ СТАЛИ
1.ЦЕЛЬ РАБОТЫ
1.Ознакомление с возможностями атомно-эмиссионного спектрального анализа с фотоэлектрической регистрацией спектра.
2.Ознакомление с работой атомно-эмиссионного квантометра на примере проведения квантометрического анализа легированной стали.
2.ОСНОВНЫЕ ТЕОРЕТИЧЕСКИЕ ПОЛОЖЕНИЯ
Эмиссионный спектральный анализ проводится с визуальной, фотографи-
ческой и фотоэлектрической регистрацией спектра.
Разработка и применение фотоэлектрических методов регистрации спектра была вызвана непрерывно возрастающими требованиями к скорости и точности анализа в металлургической и машиностроительной промышленности. Фотографические методы эмиссионного спектрального анализа уже не могли обеспечить требуемой скорости определения, так как для получения результата анализа необходимо сфотографировать спектр, проявить фотопластинку и провести фотометрирование, то есть измерение степени почернения линий анализируемых элементов. Поэтому для определения четырех – пяти элементов требуется не менее 15…25 мин. Разработка фотоэлектрических способов регистрации интенсивности излучения позволила исключить эти длительные операции и провести непосредственное измерение интенсивности излучения линий аналитической пары по величине электрического сигнала, получаемого с фотоэлектрического приемника. Это сократило время определения для десяти элементов в одном образце примерно до 2 минут.
Фактически спектральная аппаратура с фотографической и фотоэлектрической регистрацией спектра отличается только регистрирующей частью. Свет после диспергирующего элемента (вогнутая дифракционная решетка) через выходные щели попадает на фотоэлемент или фотоумножитель, который соединен с накопительным конденсатором и регистрирующим потенциометром (рис. 2). Для проведения анализа по нескольким элементам излучение аналитических линий определяемых элементов можно либо последовательно выводить на
141
одну щель по специальной программе (одноканальный прибор – стилометр или монохроматор), либо устанавливать в приборе много выходных щелей с индивидуальными приемниками излучения и одновременной регистрацией энергии для всех аналитических линий (многоканальный прибор – кванто-
метр или полихроматор).
.
4
Рис. 2. Принципиальная схема квантометра:
1 – входная щель; 2, 3, 4, 5 – плоские зеркала; 6 – откидное зеркало; 7 – вогнутая дифракционная решетка; 8 – плоскость круга Роуланда; 9 – выходная щель; 10 – вогнутое зеркало; 11 – светофильтры; 12 – фотоэлектронный умножитель
Для определения содержания каждого требуемого элемента выбирают из его спектра одну аналитическую линию, не накладывающуюся на спектры сопутствующих элементов. Но возможности выбора линий в этом варианте больше, чем при фотографической регистрации, и потому часто используются спектральные линии в спектрах различных порядков решетки. При одновременной регистрации интенсивности аналитических линий нескольких элементов для каждого из них должна быть задействована своя выходная щель. Кроме того, одна щель с фиксированным приемником излучения должна быть предна-
142
значена для линии элемента сравнения. Высокая стабильность работы генератора возбуждения позволяет использовать для всех определяемых элементов одну линию сравнения, исключая необходимость подбора строго гомологических линий.
Таким образом, число выходных щелей должно быть равно числу определяемых элементов плюс одна щель для линии элемента сравнения.
Кроме того, разработка аппаратуры с фотоэлектрической регистрацией спектра позволила полностью автоматизировать элементный спектральный анализ. Управление измерительным комплексом и распечатку результатов анализа осуществляет встроенная в прибор ЭВМ.
Современные эмиссионные анализаторы способны определять одновременно десятки элементов за одно определение.
Для проведения анализа предварительно необходимо построить градуировочные графики по стандартным образцам, состав которых точно соответствует составу анализируемых образцов. Поэтому квантометры используются только для маркировочного анализа однотипной продукции. Но при этом каждый прибор может быть настроен на анализ целого ряда типов сталей и сплавов по определенному ряду программ.
В заводских лабораториях используются отечественные (типа ДФС – 10М, ДФС-36, МФС) и зарубежные квантометры английского (типа Е), американского (типа ARL) производства.
Для определения таких элементов как углерод, сера, фосфор необходимо использовать вакуумные квантометры, например, типа ARL – 31000, Е – 600.
3. ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Отливка анализируемой стали определенного размера шлифуется наждачной бумагой непосредственно перед анализом и устанавливается в источник возбуждения квантометра.
Выбор и обработка аналитических линий спектра элементов автоматизированы. Производят два параллельных измерения, найденные усредненные значения массовых долей элементов распечатываются принтером анализатора. На распечатке указываются тип программы, шифр оператора, дата и время определения.
143
4.СОДЕРЖАНИЕ ОТЧЕТА
1.Краткое изложение методики определения.
2.Оптическая схема квантометра.
3.Фрагмент распечатки результатов анализа.
4.Заключение о содержании в стали найденных микропримесей.
Литература: [1], кн. 2, с. 31, 40, [4], с. 60…65.
Вопросы для подготовки к защите лабораторной работы
1.Перечислите основные атомно-эмиссионные и атомно-абсорбционные методы анализа.
2.Какие способы регистрации спектра используются в атомно-эмиссион- ном анализе?
3.На чем основан количественный атомно-эмиссионный анализ?
4.Какие линии называются последними?
5.Что представляет собой аналитическая пара линий?
6.Какую информацию можно получить при квантометрическом анализе?
144
2. ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА (ЛАБОРАТОРНЫЕ РАБОТЫ №№ 9…10)
Электрохимические методы анализа основаны на изучении и использовании процессов, протекающих на поверхности электрода или в приэлектродном пространстве. Они включают прямые и косвенные или титровальные методы.
В прямых методах аналитическим сигналом является электрический параметр (потенциал, сила тока, сопротивление), который функционально связан с концентрацией и который может быть измерен.
Косвенные методы основаны не на точном измерении значения величины аналитического сигнала, а на измерении изменения его величины в момент окончания аналитической реакции.
Существуют различные способы классификации электрохимических методов анализа.
Программа нашего лабораторного практикума включает изучение потенциометрии и кулонометрии. На совместном использовании этих методов основана работа кулонометрических анализаторов для определения содержания углерода и серы в объектах металлургического производства.
Определение углерода, серы, фосфора в атомно-эмиссионном спектральном анализе возможно в вакуумной области спектра, что требует использования специальных вакуумных квантометров. Возможно рентгенофлуоресцентное определение этих элементов. При анализе углеродистых сталей определения этих элементов составляют более 60% всех анализов. Кроме того, очень часто определение этих элементов необходимо по ходу плавки. Во многих случаях экономически более целесообразным оказывается определение углерода и серы с помощью кулонометрического анализатора, а определение фосфора − фотометрическим методом.
Поскольку, как уже было указано, кулонометрический анализатор представляет собой пример гибридного использования электрохимических методов анализа, а именно потенциометрии и кулонометрии, то для понимания его работы необходимо последовательно разобраться в обоих этих методах.
2.1. Потенциометрия и потенциометрическое титрование
Потенциометрический метод основан на измерении разности потенциалов между двумя электродами, погруженными в анализируемый раствор. В состав
145
установки входит индикаторный электрод, электрод сравнения и прибор для измерения потенциалов. Потенциал индикаторного электрода зависит от концентрации определяемого иона в растворе, а потенциал электрода сравнения не чувствителен к этой концентрации. Поэтому ЭДС гальванического элемента, состоящего из этих двух электродов, зависит только от концентрации определяемого иона в растворе.
Потенциометрический анализ включает прямую потенциометрию и потенциометрическое титрование.
Для измерения потенциала существуют два способа: компенсационный и некомпенсационный.
2.1.1. Измерение потенциала
Возникающую в гальваническом элементе ЭДС нельзя измерить с помощью обычного постоянно-токового вольтметра, потому что для приведения его в рабочее состояние требуется значительный ток. Но для получения истинного значения потенциала элемента через него должен протекать только ничтожно малый ток. Этим требованиям удовлетворяет потенциометр, прибор, основанный на использовании компенсационной схемы измерения разности потенциалов, в которой измеряемая ЭДС уравновешивается известной ЭДС источника постоянного тока (стандартного элемента). Принцип работы этой схемы упрощенно показан на рис.3.
1
2
3
4
Рис.3. Принципиальная схема компенсационного измерения ЭДС:
1 - источник постоянного тока; 2 - реохорд со скользящим контактом; 3 - гальванометр; 4 - электродная пара
Сущность метода заключается в том, что ЭДС исследуемого элемента компенсируется противоположно направленной известной по величине ЭДС. Полной компенсации добиваются с помощью скользящего контакта реохорда и
146
фиксируют этот момент с помощью гальванометра по отсутствию тока в цепи. Для предварительной калибровки реохорда используют элемент Вестона.
В тех случаях, когда исследуемый гальванический элемент имеет очень высокое сопротивление, порядка нескольких сотен мегаом, ток настолько слаб, что гальванометр не фиксирует его. Так происходит, например, при использовании в элементе стеклянного электрода, сопротивление которого порядка 100 МОм. В подобных случаях для измерения ЭДС пользуются либо потенциометрами с электронным усилителем, либо некомпенсационным методом измерения ЭДС. Это, в частности , относится к приборам заводского изготовления для измерения рН со стеклянным электродом, называемых рН-метрами. Они могут работать как по компенсационной схеме с электронным усилителем, усиливающим ток на несколько порядков, так и на основе электронных вольтметров прямого отсчета, снабженных электронными усилителями для получения сигнала. Шкалы приборов обоего типа калибруются как в милливольтах, так и в единицах рН. рН-метры типа потенциометров обеспечивают большую точность, чем большинство приборов прямого отсчета.
Кроме использования в конструкции рН-метров некомпенсационная схема измерения ЭДС широко распространена в практике заводских лабораторий для массовых определений при использовании потенциометрического титрования. Этот метод очень прост в аппаратурном отношении, но пользоваться им можно только в работах, не требующих большой точности.
Установка включает: источник тока, переменное сопротивление, ячейку с анализируемым раствором и парой электродов, микроамперметр. В момент измерения через раствор проходит электрический ток. Это вызывает поляризацию электродов; концентрация определяемых ионов в приэлектродном пространстве изменяется, а продукты реакции могут отлагаться на электродах. Это приводит к искажению результатов определения.
2.1.2. Электроды в потенциометрическом анализе Электроды сравнения − это полуэлементы, потенциал которых извес-
тен, постоянен и не зависит от состава анализируемого раствора. В качестве электродов сравнения используют каломельный и хлоридсеребряный электроды.
Индикаторные электроды могут быть металлическими и мембран-
ными.
Металлические электроды изготавливают из проволоки, пластинок или
147
цилиндрических болванок. Они могут быть инертными и активными. Инертные электроды, изготовленные из таких металлов, как золото и
платина, используются в окислительно-восстановительных реакциях. Потен-
циал таких электродов является функцией соотношения концентраций (активностей) окисленной и восстановленной формы одного или нескольких веществ в растворе.
Активные электроды - это электроды I рода: серебряный, медный, кадмиевый, то есть металл, способный давать обратимые полуреакции. Для активных металлических электродов потенциал этого электрода отражает активность ионов металла в растворе. Например, для серебряного электрода, на поверхности которого протекает реакция:
Ag+ +e Ag ,
E EAg0 +/Ag 0,059n lg aAg .
Непригодными для этой цели оказываются железо, никель, вольфрам, хром, кобальт, так как их потенциалы невоспроизводимы вследствие кристаллической деформации в их структурах и образования оксидных слоев на поверхности.
Металлические электроды могут служить в качестве индикаторных и по отношению к анионам, образующим труднорастворимые соединения с соответствующим катионом. Например, серебряный электрод может использоваться для определения содержания ионов серебра и для определения галогенидионов, образующих труднорастворимые соединения с серебром.
Мембранные электроды в зависимости от материала мембраны подразделяются на несколько категорий:
1)стеклянные электроды;
2)электроды с жидкими мембранами;
3)электроды с твердыми или осадочными мембранами;
4)электроды с газочувствительными мембранами;
5)ферментные электроды.
Все мембранные электроды являются ионоселективными. По определению ИЮПАК (Международный союз теоретической и прикладной химии - International Union of Pure and Applied Chemistry - IUPAC), "ионоселективные
148
электроды − это сенсоры (чувствительные элементы, датчики), потенциалы которых линейно зависят от lg a определяемого иона в растворе".
Полупроницаемая мембрана – это тонкая пленка, отделяющая внутреннюю часть электрода от анализируемого раствора и способная пропускать преимущественно ионы только одного вида.
Классификация ионоселективных электродов в соответствии с рекомендациями ИЮПАК:
-первичные ионоселективные электроды;
-электроды с подвижными носителями;
-сенсибилизированные (активированные) электроды.
Эти электроды характеризуются тремя основными параметрами: элек-
тродная функция, селективность и время отклика.
Электродная функция. Электродная мембрана контактирует с двумя растворами: анализируемым и внутренним. На обеих сторонах мембраны происходит обмен ионами с ионами обоих растворов. Поскольку активности ионов в растворе и в мембране различаются, то на обеих сторонах мембраны возникают граничные потенциалы Е1 и Е2. Если и во внешний, и во внутренний растворы поместить электроды сравнения, то можно измерить разность потенциалов Е1 и Е2, называемую мембранным потенциалом Ем
Ем = E1 - E2 = 0,059 lg a1 / a2 ,
где а1 и а2 - активности ионов А+ в анализируемом и во внутреннем растворах. Поскольку активность ионов во внутреннем растворе постоянна, потенциал мембранного электрода фактически линейно зависит от активности ио-
нов А+ в анализируемом растворе:
Ем = const + 0,059 lg a1.
Линейность функции сохраняется в определенном интервале активностей, определяемом природой мембраны. Точка перегиба на графике характеризует величину предела обнаружения.
Селективность. В идеальном случае электродная функция мембранного электрода зависит только от активности определяемых ионов Мz+. Однако на практике на величину потенциала наряду с определяемым влияют и другие ионы, присутствующие в растворе и, следовательно, частично тоже проходящие через мембрану. Селективность (избирательность) электрода зависит от двух основных факторов: способности ионов мембраны М1 обмениваться с посто-
149
ронними ионами раствора М2, а также от соотношения подвижностей этих ионов в мембране. Зависимость потенциала от активности определяемого и сопутствующего ионов выражается уравнением
E = E0 + 0.059 lg (a (M1) + K(M1 /M2 ) · a (M2 )), n
где К − коэффициент селективности, определяющий вклад посторонних ионов М2 в возникновение электродного потенциала. Чем меньше этот коэффициент, тем выше селективность электрода. Если величина коэффициента селективности значительно меньше единицы, то такой электрод называют ионоселективным. Например, если для натриевого электрода величина К (Na+/K+) = 3,6·10-4, то это означает, что при измерении потенциала электрода раствор с концентрацией натрия, равной 3,6·10-4 моль/л, будет давать то же значение потенциала, что и раствор калия с концентрацией, равной 1 моль/л.
Время отклика - это время, требуемое на измерение концентрации раствора, которое определяют по изменению величины потенциала электрода с момента погружения его в раствор. В зависимости от природы мембраны время отклика меняется от нескольких секунд до нескольких минут. Этот параметр необходимо учитывать при использовании электрода для непрерывных измерений в потоке или при автоматизированных измерениях.
Стеклянные электроды
Стеклянный электрод для измерения рН имеет наибольшую известность и практическое применение. Удобство в работе и сравнительно меньшая зависимость от влияния посторонних примесей позволили ему практически полностью вытеснить все другие электроды, использовавшиеся ранее для тех же целей. Он применяется для определения рН не только истинных растворов, но и вязких, и полутвердых жидкостей; его можно использовать в присутствии сильных окислителей и восстановителей, белковых веществ и газов.
Он изготавливается припаиванием тонкого наконечника, чаще всего в форме шарика, из рН-чувствительного стекла к концу толстостенной стеклянной трубки. рН-чувствительная мембрана имеет толщину от нескольких сотых до 0,1 мм. Наиболее широко используется стекло марки "корнинг 015" приблизительного состава: 22 % оксида натрия, 6 % оксида кальция, 72 % оксида кремния. Внутрь трубки заливают раствор децимолярной хлороводородной кислоты, в который погружают хлоридсеребряный электрод. Внешний вывод его подсоединяют к одному из полюсов прибора для измерения рН. Каломельный
150
(или второй хлоридсеребряный) электрод присоединяют к другому полюсу ячейки. Потенциалы обоих электродов сравнения (внешнего и внутреннего) не зависят от рН и сохраняют свое значение во время измерения рН. Постоянен и потенциал асимметрии. Поэтому зависимость потенциала ячейки от активности ионов водорода выражается уравнением
E = K + 0,059 lg α (H+).
Для того чтобы стеклянная мембрана функционировала как рН − электрод, она должна быть предварительно гидратирована. С этой целью электрод предварительно выдерживается в воде или в сантимолярном растворе хлороводородной кислоты. В процессе гидратации адсорбируется примерно 50 мг воды на кубический сантиметр стекла. При этом происходит обмен между катионами натрия стекла и протонами раствора. В результате поверхность гидратированной стеклянной мембраны состоит почти исключительно из кремниевой кислоты. Слой ее геля достигает толщины 10-4 − 10-5 мм. На внешней стороне геля все пустоты заняты ионами водорода. При движении внутрь геля ионы водорода все больше замещаются ионами натрия.
Стеклянный рН -чувствительный электрод обладает высокой специфичностью по отношению к ионам водорода в интервале рН от 2 до 9. Ошибка измерения в сильнокислых растворах, предположительно связана с разрушением стекла. В щелочных растворах селективность по отношению к щелочным металлам уменьшается, и электрод начинает реагировать не только на ионы водорода, но и на ионы калия, натрия и других щелочных металлов.
Промышленностью выпускаются сравнительно недорогие электроды, различные по форме и размерам. Наряду с наиболее распространенными погруженными электродами выпускаются специальные электроды, например для измерения рН в капле, в потоке движущейся жидкости; микроэлектроды для физиологических исследований, позволяющие измерять рН внутри живой клетки, в полости зуба, в желудке и т.д.
Изменяя состав стекла, можно получать мембраны, обладающие высокой селективностью по отношению к ионам металлов и пониженной − по отношению к ионам водорода (табл. 3).
151

|
|
Таблица 3 |
|
Состав и рабочие характеристики стеклянных электродов |
|||
|
|
|
|
Опреде- |
Состав стеклянной |
Коэффициент селективности и до- |
|
ляемый |
|||
мембраны |
полнительные характеристики |
||
ион |
|||
|
|
||
|
|
K(Na+,K) = 3,6·10-4 при |
|
Na+ |
Na2O - Al2O3-SiO2 |
||
|
|
Cmin(Na+) = 10-5моль/л |
|
|
|
K(Na+,K+) = 10-5, показания неустойчи- |
|
Na+ |
Li2O - Al2O3 -SiO2 |
||
|
|
вы во времени |
|
|
|
K(Na+ ,K+) =10-3 |
|
Na+ |
Li2O - B2O3 -SiO2 |
||
|
|
K(K+ ,Na+) =0,05 при |
|
K+ |
Na2O - Al2O3-SiO2 |
||
|
|
Cmin (K+) = 10-4моль/л |
|
Об остальных типах электродов подробно см. [2].
Потенциометрия и потенциометрическое титрование основаны на использовании известного Вам уравнения Нернста, связывающего величину потенциала электрода с активностями окисленной и восстановленной форм окис-
лительно-восстановительной системы |
|
|
|
|
|
||
E E0 |
0.059 lg |
aox |
E0 |
0.059 lg |
[ox] ox |
. |
|
a |
|
||||||
|
n |
|
n |
[red] |
red |
||
|
|
red |
|
|
|
||
Поскольку абсолютное значение потенциала измерить невозможно, измеряется разность потенциалов системы, один из электродов которой имеет постоянное значение потенциала – электрод сравнения, а потенциал второго электрода – индикаторного – зависит от концентрации (активности) определяемого иона.
В качестве электрода сравнения независимо от типа реакции используется хлоридсеребряный электрод (рис. 4), а тип индикаторного электрода подбирается в зависимости от изучаемой реакции.
152

Рис. 4. Хлоридсеребрянный электрод
1 – серебряная проволока; 2, 4 – внутренний раствор KCl (нас.); 3 – отверстие для контакта; 5 – асбестовое волокно
При определении содержания углерода в сталях и сплавах, как будет видно дальше, мы будем контролировать изменение концентрации ионов водорода (или величины рН). Поэтому в качестве индикаторного электрода мы бу-
дем использовать стеклянный электрод с рН – функцией (рис. 5).
Рис. 5. Стеклянный электрод:
1 – стеклянная рН - чувствительная мембрана; 2 – 0,1 М раствор HCl, насыщенный AgCl; 3 – серебряная проволочка; 4 – стеклянная трубка
Потенциал стеклянного электрода определяется уравнением:
E const 0.059lg aH .
153
2.2. Кулонометрия
Электрохимический метод анализа, основанный на измерении точного количества электричества, пропущенного через электролизер, называют кулонометрическим.
Кулонометрия основана на использовании законов Фарадея для определения количества вещества, которое образовалось или прореагировало при протекании электродной реакции:
1.Количество вещества, электропревращенного в процессе электролиза (восстановленного или окисленного), прямо пропорционально количеству электричества, протекшего через электролит.
2.При протекании одинакового количества электричества через разные электролиты выделяются или растворяются количества веществ, прямо пропорциональные их электрохимическим эквивалентам.
Необходимым условием кулонометрического определения является единичный выход по току: все прошедшее через электролизер электричество затрачивается на выделение определяемого элемента.
Масса электрохимически окисленного или восстановленного вещества может быть вычислена по формуле
m = MQ / nF ,
где m – масса вещества, выделившегося в ходе электролиза, г; М – молярная или атомная масса, г·моль-1 ; Q – измеренное количество электричества, Кл, равное произведению силы тока на время Q = I·t (1 Кл = 1 А·с); n – число электронов; F – число Фарадея (электрохимический эквивалент моля), равное 96485 Кл·моль-1.
Как видно из приведенного уравнения, кулонометрические определения являются безградуировочными и не требуют стандартных образцов.
154

ЛАБОРАТОРНАЯ РАБОТА №9
ПОТЕНЦИОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ КИСЛОТЫ
1.ЦЕЛЬ РАБОТЫ
1.Ознакомление с методом прямой потенциометрии и потенциометрического титрования.
2.Потенциометрическое титрование хлороводородной кислоты.
2.ОСНОВНЫЕ ТЕОРЕТИЧЕСКИЕ ПОЛОЖЕНИЯ
Потенциометрия основана на измерении электрохимического потенциала
обратимого по отношению к определяемому иону индикаторного электрода относительно электрода сравнения, потенциал которого не изменяется в процессе определения. Таким образом электрод сравнения служит уровнем отсчета потенциала индикаторного электрода. Теоретические принципы потенциометрии основаны на известном уравнении Нернста. По способу реализации различают прямую потенциометрию и потенциометрическое титрование.
Метод прямой потенциометрии обладает важными достоинствами:  В процессе измерения анализируемый раствор не разлагается.
В процессе измерения анализируемый раствор не разлагается.
 Метод позволяет проводить определения, используя очень малые объемы анализируемого раствора (микро- и даже нанолитры).
Метод позволяет проводить определения, используя очень малые объемы анализируемого раствора (микро- и даже нанолитры).
Результаты измерения получают непосредственно в виде электрического сигнала, что позволяет использовать прямую потенциометрию для непрерывного и динамического контроля изучаемого процесса с помощью ионселективных электродов.
155
3. ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Для измерения ЭДС гальванических элементов с использованием ионоселективных электродов(ИСЭ) используются электронные вольтметры с высоким входным сопротивлением (иономеры, рН-метры).
Мы будем использовать отечественный автотитратор или блок автоматического титрования БАТ-15 , который выпускается в комплекте с бюреткой и рН-метром – милливольтметром, имеющим диапазон измерений от -1 до +14 (1000…1400 мВ)
Схема установки приведена на рис. 6.
Рис. 6. Схема установки для автоматического титрования
1- бюретка с титрантом; 2 – магнитный клапан; 3 – рН – метр с двумя электродами: индикаторным и сравнения; 4 – блок автоматического титрования БАТ – 15;5 - реле электромагнитного клапана; 6 – электронный усилитель; 7 - задатчик конечной точки титрования; 8 – ячейка с титруемым раствором
156
Проведение определения
Прямая потенциометрия (измерение рН)
Подключить прибор к сети для прогрева в течение 30 мин. Затем проверить настройку прибора по двум буферным растворам – в кислой и в щелочной областях рН. Для этого залить в чистый сухой стакан небольшое количество буферного раствора с известным значением рН и замерить полученное значение величины рН. При необходимости скорректировать полученную величину с помощью специальной настройки. Перед измерением второго раствора электроды снова тщательно промыть дистиллированной водой и осторожно удалить избыток воды фильтровальной бумагой.
Затем получить у преподавателя в стакан для измерений испытуемый раствор и измерить его значение рН. Записать показания в рабочий журнал.
Потенциометрическое титрование хлороводородной кислоты
Получить у преподавателя испытуемый раствор в стакан для титрования, поставить стакан на площадку магнитной мешалки и опустить в него мешалку. Погрузить в испытуемый раствор два электрода: индикаторный (стеклянный) и электрод сравнения (хлоридсеребряный). Электроды должны быть предварительно обмыты дистиллированной водой и обсушены фильтровальной бумагой. Включить мешалку.
Заполнить бюретку 0,1 М раствором гидроксида натрия, включить режим автоматического титрования и открыть кран бюретки. В начале процесса титрования поступление рабочего раствора в ячейку идет с заданной нами скоростью. Но по мере приближения к заданной точке конца титрования рН = 7 скорость подачи титранта в ячейку автоматически замедляется регулировочным магнитным клапаном, открывающим или закрывающим подачу титранта. В момент достижения заданной точки конца титрования рН = 7 подача титранта автоматически прекращается. Объем титранта V(NaOH), затраченный на проведение титрования, отсчитывают по показанию шкалы бюретки.
По значению известной величины молярной концентрации эквивалента гидроксида натрия с(NaOH) рассчитываем T(NaOH /HCl):
T(NaOH/HCl) = [ñ(NaOH/1000] M(HCl).
157
Теперь можем вычислить содержание HCl в анализируемом растворе m(HCl)
m(HCl) = T(NaOH/HCl) V(NaOH).
4. СОДЕРЖАНИЕ ОТЧЕТА
Отчет должен содержать:
1.Цель работы.
2.Схематические рисунки индикаторного электрода и электрода сравне-
ния.
3.Результаты измерения рН в контрольном растворе.
4.Результаты титрования анализируемого раствора хлороводородной кислоты и расчет ее количества.
Литература: [1], кн. 2, с.189…207.
Вопросы для подготовки к защите лабораторной работы
1.Перечислите основные типы ионоселективных электродов.
2.Какие требования предъявляются к электродам сравнения?
3.Какое уравнение связывает величину измеренного потенциала и концентрацию определяемого иона?
158
ЛАБОРАТОРНАЯ РАБОТА №10 ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ УГЛЕРОДА В СТАЛЯХ НА
КУЛОНОМЕТРИЧЕСКОМ АНАЛИЗАТОРЕ
1.ЦЕЛЬ РАБОТЫ
1.Ознакомление с работой кулонометрического анализатора на углерод.
2.Определение массовой доли углерода в стали.
2.ОСНОВНЫЕ ТЕОРЕТИЧЕСКИЕ ПОЛОЖЕНИЯ
Внастоящее время для определения содержания углерода, серы или одновременного определения содержания обоих элементов в металлах выпускаются экспресс-анализаторы двух типов: на основе кулонометрического метода анализа и на основе анализа продуктов сжигания методом инфракрасного поглощения. На рынке аналитического приборостроения имеется продукция отечественного и зарубежного производства. Наибольшее распространение имеют анализаторы типов АН – 8012, 8112; АС – 8032 , 8132; АУС – 8044, 8144 и др.
Внастоящей работе рассмотрено определение содержания углерода на основе работы кулонометрического анализатора.
Метод основан на сжигании навески пробы в токе кислорода при температуре 1250-1350 оС, поглощении образовавшегося диоксида углерода поглотительным раствором с определенным начальным значением рН и последующим кулонометрическим титрованием образовавшейся сильной кислоты. Количество электричества, необходимое для этого, пропорционально массовой доле углерода в навеске пробы. Схема кулонометрического анализатора представлена на рис. 7.
159

Рис. 7. Схема кулонометрического анализатора на углерод
1 – трубчатая печь, в которую помещена лодочка с навеской; 2 – электролитическая ячейка; 3 – поглотительный раствор с рН 10,2; 4 – индикаторный стеклянный электрод; 5 – хлоридсеребряный электрод сравнения ; 6 – рН-метр; 7 – регулятор включения генераторного тока; 8 – источник генераторного тока; 9 – катод; 10 – цинковый анод; 11 – проницаемая перегородка; 12 – вспомогательный раствор, содержащий хлорид калия и ферроцианид калия; 13 – интегратор тока.
160
Основные реакции, протекающие в ходе определения
При сжигании навески металла в печи углерод окисляется до диоксида и по трубке поступает в электролитическую ячейку, где образуется угольная кислота:
C O2 CO2
H2O + CO2 H2CO3
Угольная кислота является слабым электролитом, диссоциирует очень незначительно. Поэтому в состав поглотительного раствора вводится хлористый стронций, при взаимодействии с которым образуется эквивалентное количество сильной хлороводородной кислоты:
SrCl2 + H2CO3 SrCO3 + 2HCl
HCl H+ + Cl-.
В результате в поглотительном растворе происходит резкое повышение концентрации ионов водорода, на что реагирует индикаторный стеклянный электрод. Это приводит к изменению силы тока на выходе усилителя рН – метра, что приводит к включению источника генераторного тока. При этом на катоде начинает выделяться водород
2H+ + 2e- H2 ,
а цинковый анод растворяется
Zn - 2e- Zn2+.
Избыточные ионы цинка взаимодействуют с ферроцианидом калия и осаждаются:
K4[Fe(CN)6 ] + Zn2+ K2Zn3[Fe(CN)6 ]2 .
Эти процессы протекают до тех пор, пока стеклянный электрод не зафиксирует снижение концентрации ионов водорода до первоначального значения. В этот момент цепь разомкнется и прекратится генерирование тока. Интегратор тока зафиксирует количество электричества, затраченное на выделение водорода, которое определяется количеством углерода, содержащимся в анализируемом образце. Массовая доля углерода будет указана на цифровом табло.
3.ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Воснову используемой методики определения содержания углерода в
сталях положен ГОСТ 22536-87.
161
Перед началом работы для удаления следов углерода через разогретый анализатор пропускают кислород и прокаливают трубку. При анализе пробы с массовой долей углерода менее 0,10 % необходима дополнительная очистка га- за-носителя от диоксида серы. Для этого в фильтр-поглотитель, находящийся между поглотительным сосудом и печью, помещают диоксид марганца или гидроперит. Допускается использовать свинец для устранения влияния серы при ее массовой доле менее 0,03 %.
Навеску пробы около 1,000 г помещают в фарфоровую лодочку и покрывают слоем плавня. В качестве плавня используют медь по ГОСТ 546-79, олово по ГОСТ 13610-79 или оксиды этих металлов. Соотношение масс плавня и пробы – 0,5:1 или 1:1. При анализе углеродистых сталей допускается проводить сжигание навески пробы без плавня. Лодочку с навеской и плавнем помещают в рабочую часть печи и сжигают пробу в токе кислорода при температуре 1250-1350 оС.
Анализ считают законченным, если показания прибора не изменяются в течение 1 минуты.
Массовую долю углерода ω(С) определяют по цифровому табло анализатора за вычетом результата контрольного опыта:
ω(C) = A - AK ,
где А – показания цифрового табло прибора, полученные в результате сжигания навески пробы, %; Ак – среднее арифметическое значение показаний цифрового табло прибора, полученное в результате сжигания плавня или проведения контрольных анализов, % (масс.).
4. СОДЕРЖАНИЕ ОТЧЕТА
Отчет о работе должен содержать:
1.Формулу закона Фарадея.
2.Схему кулонометрического анализатора.
3.Результаты определения и расчет массовой доли углерода в анализируемой стали.
Литература: [1], кн. 2, с.239…245; [4], с. 37…40.
Вопросы для подготовки к защите лабораторной работы
1.Сформулируйте законы Фарадея и запишите на их основе формулу для расчета массы вещества, выделенного в процессе электролиза.
2.Нарисуйте принципиальную схему кулонометрического анализатора на углерод.
3.Запишите основные реакции, протекающие в процессе определения.
162
3. РЕНТГЕНОСПЕКТРАЛЬНЫЙ АНАЛИЗ (ЛАБОРАТОРНАЯ РАБОТА № 11)
Рентгеноспектральный анализ так же, как и атомный эмиссионный спектральный анализ позволяет получить и качественную, и количественную информацию о составе анализируемых веществ. Но в отличие от атомного спектрального анализа проведение рентгеноспектрального анализа не требует разрушения (атомизации) образца, так как рентгеновское излучение и поглощение обусловлено возбуждением внутренних электронов определяемого элемента, переходы которых могут происходить как в свободном элементе, так и в элементе, входящем в состав какого-либо вещества. Поэтому характер рентгеновского атомного спектра не зависит от химического состояния определяемого элемента в образце.
В основе рентгеноспектрального анализа лежит использование зависимости частоты излучения линий характеристического спектра элементов от их атомного номера (закон Мозли) и связи между интенсивностью этих линий и числом атомов, принимающих участие в излучении.
Рентгеноспектральный анализ включает три метода: по первичным спектрам испускания, по вторичным спектрам испускания (флуоресцентный анализ), по спектрам поглощения (используется редко). Первичные спектры испускания возникают при прямой бомбардировке образца электронами больших энергий, а вторичные – при облучении атомов вещества рентгеновскими лучами.
Примером использования в анализе первичных рентгеновских лучей является электронно-зондовый рентгеновский микроанализатор. В металлургии микрозонд используют, например, для локального многоэлементного анализа поверхностных слоев образцов, содержащих микроскопические гетерофазы, например для исследования природы частиц, образующихся на границах зерен, в том числе для анализа материалов высоких технологий, для исследования состава продуктов следов коррозии.
Несмотря на то, что интенсивность вторичного излучения в тысячи раз меньше интенсивности лучей, полученных первичным методом, флуоресцентная аппаратура почти полностью вытеснила из практики установки прямого возбуждения рентгеновских лучей.
163
3.1.Рентгенофлуоресцентный анализ
Врентгенофлуоресцентном анализе исследуемая проба подвергается воздействию первичного рентгеновского излучения трубки. Характер возникающего вторичного излучения пробы зависит от качественного и количественного состава образца. При этом флуоресцентное излучение содержит только характеристические спектральные линии, в то время как на первичный эмиссионный спектр накладывается спектр непрерывного излучения.
Рентгеновский спектрометр включает рентгеновскую трубку с высокой интенсивностью излучения (чаще всего с вольфрамовым анодом мощностью до 3,5 кВт); камеру образца; коллиматор – систему параллельных молибденовых пластин, предназначенную для пропускания параллельно идущих только в одном направлении лучей; кристалл – анализатор, работающий как дифракционная решетка, и приемник излучения (пропорциональный или сцинтилляционный счетчики). Оба типа детекторов подсчитывают отдельные кванты рентгеновского излучения в виде дискретных электрических импульсов. После селекции и дискриминации электрические импульсы регистрируются суммирующим устройством и далее используются для построения калибровочных графиков или определений элементов.
Изготовить дифракционную решетку для рентгеновского излучения практически невозможно, поскольку для этого необходимо на одном миллиметре решетки нанести несколько миллионов штрихов. Поэтому в качестве дифракционной решетки используют естественные кристаллы (чаще всего фторид лития, топаз, фторид лития). Кристаллические минералы образуют систему кристаллографических плоскостей, которые и составляют своеобразную естественную дифракционную решетку.
Рентгеновские фотоны первичного излучения трубки выбивают из атома пробы электроны, которые находятся на орбитали в непосредственной близости от ядра атома. На образовавшиеся при этом вакансии переходят электроны с более отдаленных от ядра оболочек. Этот переход сопровождается возникновением вторичного рентгеновского излучения. Обозначение и классификация линий рентгеновского спектра учитывает числовые значения главного квантового числа (n = 1, 2, 3, 4), соответствующие им обозначения символами K, L, M, N. Все переходы, заканчивающиеся на K - уровне, приводят к образованию K –линий.
Индекс (K ) показывает, что переход произошел с L – оболочки на K – оболочку.
164
Переходу с M на K оболочку соответствует символ Kβ.. При переходе на L – оболочку образуются линии Lα, Lβ и т. д. Разность энергий для осуществления β - переходов всегда больше, чем для – переходов, поэтому β - линии всегда лежат в более коротковолновой части спектра по сравнению с - линиями.
В противоположность оптической спектроскопии при таких электронных переходах изменяется главное квантовое число, поэтому рентгеновские спектры отличаются от оптических малым числом линий, а, следовательно, более легкой расшифровкой.
Дифракция ретгеновских лучей в кристалле – анализаторе описывается законом Вульфа–Брэгга:
n = 2 d sin θ,
где n – целое число, показывающее порядок спектра (обычно рассматривается только спектр первого порядка); λ − длина волны; d − расстояние между соседними плоскостями кристалла; θ − угол между лучом и отражающей плоскостью кристалла.
Рентгенофлуоресцентный анализ позволяет получить информацию каче-
ственного и количественного характера. Большинство серийных приборов позволяет анализировать элементы с атомным номером больше двенадцати.
Качественный анализ основан на вычислении по уравнению ВульфаБрэгга длины волны рентгеновского излучения, соответствующего определенному электронному переходу L → K, M → K. Полученные результаты сопоставляются с данными справочных таблиц для рентгенофлуоресцентного метода, которые позволяют определить, какому элементу принадлежат эти линии.
Длины волн аналитических линий в рентгенофлуоресцентном анализе измеряются десятыми и сотыми долями нанометра. Они зависят от типа переходаи вида элемента. В зависимости от того, какой элемент необходимо определить и какова длина волны аналитической линии, подбирается кристалл–анализатор с требуемым межплоскостнымрасстоянием(кварц, топаз, фторидлитияит. д.).
Для количественного определения измеряют интенсивность рентгенофлуоресцентной линии. Она зависит от концентрации данного элемента в пробе, то есть от числа излучающих атомов, а также от интенсивности и длины волны
165
первичного рентгеновского излучения, от толщины пробы, от природы и содержания сопутствующих элементов (матричный эффект).
Для коррекции влияния матричного эффекта анализируемую пробу необходимо сравнивать с близкими по химическому составу стандартными пробами. Предварительную калибровку выполняют по нескольким эталонным образцам методом калибровочного графика, как правило, с использованием стандартных образцов предприятия, либо методом внутреннего стандарта. Кроме того, в отличие от оптической спектроскопии влияние сопутствующих элементов на интенсивность линий корректируется и расчетным путем, методом фундаментальных параметров.
В противоположность оптической эмиссионной спектроскопии рентгенофлуоресцентный анализ наряду с определением следовых количеств позволяет определять высокие содержания основных компонентов со среднеквадратичной погрешностью в пределах от 2…5 % отн. Поэтому в практике работы заводских лабораторий он является ценным дополнением методов оптической спектроскопии при определении основных компонентов.
Рентгенофлуоресцентным методом можно определять элементы, начиная с магния и кончая ураном. Для более легких элементов выход флуоресценции составляет менее 0,1 %, поскольку энергия, высвобождающаяся при осуществлении внутриатомных переходов, расходуется на возбуждение и эмиссию внешних валентных электронов.
Анализируемые вещества могут находиться в твердом, жидком, порошкообразном состоянии. Они могут быть нанесены на поверхность или осаждены на фильтр. Поверхность металлических образцов должна быть плоской и гладкой, поскольку интенсивность излучения зависит от шероховатости поверхности. Стружка или мелкие кусочки прессуются в плоские таблетки, порошок прессуют в брикеты или таблетки с бурой. При проведении анализа образец вращают, чтобы уменьшить вариации сигнала, связанные с неоднородностями поверхности образца.
Представленные на современном рынке аналитического приборостроения рентгенофлуоресцентные спектрометры полностью автоматизированы. Оператор подготавливает образцы, устанавливает их в спектрометр и запускает измерения. Всем дальнейшим процессом управляет персональный компьютер. Программное обеспечение для проведения качественного и количественного анализов многоэлементных веществ в соответствии с требованиями норматив-
166
ных документов, регламентирующих точность анализа (ГОСТ, ASTM, ISO), прилагается. Это позволяет проводить анализ входного сырья, продуктов технологического процесса и конечной продукции в черной, цветной и вторичной металлургии: анализ чугунов, высоколегированных и низколегированных сталей, железорудного сырья и ферросплавов, шлаков, сплавов на основе магния, алюминия, меди. В области металлургии имеется ряд аттестованных методик.
Таким образом, основными преимуществами рентгенофлуоресцентного анализа являются возможность проведения анализа независимо от агрегатного состояния веществ, без разрушения образца; широкий диапазон определяемых содержаний от 10-4 до 100 %; высокая точность анализа; экспрессность; относительная простота спектров и удобство их расшифровки.
167
ЛАБОРАТОРНАЯ РАБОТА №11 ЭЛЕМЕНТНЫЙ АНАЛИЗ СТАЛИ РЕНТГЕНОФЛУОРЕСЦЕНТНЫМ
МЕТОДОМ
1.ЦЕЛЬ РАБОТЫ
1.Ознакомление с методом рентгенофлуоресцентного анализа.
2.Элементный анализ легированной стали рентгенофлуоресцентным методом.
2.ОСНОВНЫЕ ТЕОРЕТИЧЕСКИЕ ПОЛОЖЕНИЯ
Метод основан на зависимости интенсивности характеристических линий элемента от его массовой доли в пробе. Флуоресценция возбуждается первичным рентгеновским излучением, источником которого служит трубка с вольфрамовым анодом. Излучение элементов разлагается в спектр с помощью кри- сталла-анализатора. Интенсивность характеристических линий флуоресценции элементов измеряется ионизационным детектором и используется для построения калибровочных графиков. Массовая доля определяемого элемента определяется с помощью градуировочных характеристик, построенных по стандартным образцам.
3. ПОРЯДОК ВЫПОЛНЕНИЯ РАБОТЫ
Элементный анализ легированной стали проводится на основе ГОСТ
28033-89.
Анализируемая проба обрабатывается механически до заданных размеров. Поверхность для анализа шлифуется наждачной бумагой, тип 2, затем протирается этиловым спиртом и устанавливается в камеру рентгеновской трубки. Производится 2…4 измерения. Одновременно можно определять до нескольких десятков элементов. Например, при использовании анализатора PW – 16000 можно определить до 32 элементов в образцах легированной стали. В их числе: фосфор, марганец, хром, никель, кобальт, медь, молибден, вольфрам, ванадий, титан и ниобий.
4. СОДЕРЖАНИЕ ОТЧЕТА
Отчет о работе должен содержать:
1.Изложение методики определения.
2.Схему рентгенофлуоресцентного анализатора.
3.Фрагмент распечатки результатов анализа.
168
Литература: [1], кн. 2, с. 110…126; [4], с. 60…69
Вопросы для подготовки к защите лабораторной работы
1.Перечислите основные виды ренгеноспектрального анализа.
2.Запишите уравнение закона Вульфа – Брэгга и разъясните значение входящих в него параметров.
3.Как проводится качественный и количественный рентгенофлуоресцентные анализы?
4.Каковы основные достоинства рентгенофлуоресцентного анализа и его основное преимущество по сравнению с эмиссионным спектральным анализом?
169