

- •1. ИНФОРМАЦИЯ О ДИСЦИПЛИНЕ
- •2. РАБОЧИЕ УЧЕБНЫЕ МАТЕРИАЛЫ
- •2.2. Тематический план дисциплины
- •2.3. Структурно-логическая схема дисциплины3
- •2.5. Практический блок
- •2.6. Балльно-рейтинговая система оценки знаний
- •3. ИНФОРМАЦИОННЫЕ РЕСУРСЫ ДИСЦИПЛИНЫ
- •3.1. Библиографический список
- •3.2. Опорный конспект
- •РАЗДЕЛ 1. ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
- •3.3. Глоссарий
- •3.4. Технические и программные средства обеспечения дисциплины
- •3.5. Методические указания к выполнению лабораторных работ
- •4. БЛОК КОНТРОЛЯ ОСВОЕНИЯ ДИСЦИПЛИНЫ
- •4.1. Общие указания
- •ПРИЛОЖЕНИЯ
ПРИЛОЖЕНИЯ
260
Приложение 1
Классификация видов анализа
261
|
|
Приложение 2 |
|
Виды анализа по природе определяемых частиц |
|||
|
|
|
|
Вид анализа |
Установление наличия и |
Примеры |
|
содержания |
|
||
|
|
|
|
|
|
|
|
Элементный |
Отдельных элементов |
Fe, Cr, Mn, C, S, O, N, Al, Si |
|
|
|
|
|
Молекулярный |
Молекул, соединений |
Al2O3, SiO2 - í åô åëèí |
|
|
|
FeO, Fe2O3 - железн ая руда |
|
|
|
FeS2 - ï èðèò |
|
|
|
CO, CO2 , N2 âî çäóõ |
|
Фазовый |
Отдельных фаз анализи- |
Карбиды на основе железа, |
|
|
руемого объекта |
хрома, нитриды в стали |
|
|
|
|
|
Функциональный |
Функциональных групп в |
Карбоксильных –СООН, |
|
|
молекулах органических |
амино- -NH2 , - NO - |
|
|
соединений |
нитрозо |
|
|
|
|
|
Приложение3
Виды анализа в зависимости от количества вещества, использованного для анализа
Вид анализа |
Масса пробы, г |
Объем раствора, мл |
|
|
|
Макроанализ |
..>0,1 |
10 103 |
Полумикроанализ |
0,01 – 0,1 |
10 1 10 |
Микроанализ |
<0,01 |
10 2 1 |
Субмикроанализ |
10 4 10 3 |
10 2 |
Ультрамикроанализ |
10 4 |
10 3 |
262
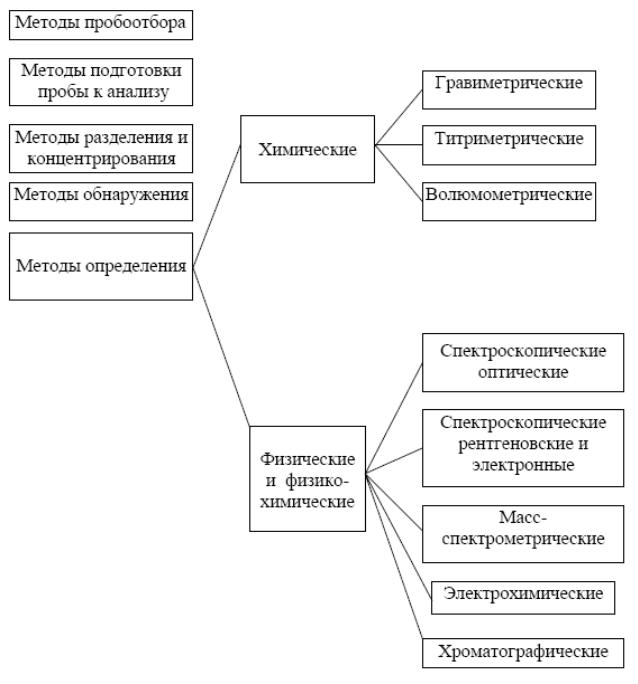
Приложение 4
Методы аналитической химии
263
Приложение 5
Качественный химический анализ
Качественный анализ неорганических и органических веществ предполагает идентификацию компонентов анализируемого объекта, то есть установле-
ние качественного состава вещества или смеси веществ. Количественный анализ устанавливает содержание или концентрации составных частей анализируемых веществ.
Химические реакции обнаружения можно выполнять «мокрым» и «сухим» путем. Чаще используется первый вариант. В этом случае анализируемый объект предварительно необходимо растворить в воде, кислоте или щелочи. При невозможности прямого растворения используют сплавление с последующим выщелачиванием.
Все аналитические реакции сопровождаются внешним эффектом:
образование или растворение осадка;
появление или изменение окраски;
выделение газов.
Каждая из используемых в качественном анализе реакций является реакци-
ей идентификации с той или иной степенью селективности или избирательности.
На использовании групповых реагентов основан систематический качественный анализ. С их помощью последовательно разделяют катионы исследуемого образца на аналитические группы, используя реакции осаждения. Деление катионов на аналитические группы не совпадает с делением элементов на группы в Периодической системе элементов.
Воснове аналитической классификации на группы лежит одинаковое взаимодействие с групповым реагентом.
Например, HCl образует осадки с ионами Ag(I), Hg(I), Tl(I), Pb(II); H2SO4- с ионами Ca(II), Sr(II), Ba(II), Pb(II).
Следовательно, HCl является групповым реагентом для катионов Ag(I), Hg(I), Tl(I), Pb(II); H2SO4 - для катионов Ca(II), Sr(II), Ba(II), Pb(II).
Вкачестве групповых реагентов могут использоваться сероводород, сульфид аммония, гидроксид аммония в зависимости от принятой схемы систе-
матического хода анализа. Последовательное разделение смеси веществ на группы, подгруппы позволяет подойти к дробному определению каждого иона
спомощью специфических реакций.
264

Метод дробного анализа, основанный на использовании специфических реагентов, без предварительного разделения на группы возможен с помощью маскирования мешающих элементов, изменения рН и других условий.
Каждая аналитическая группа имеет свой групповой реагент – осадитель, который одновременно образует необходимые соединения (не обязательно осадки) со всеми катионами аналитической группы. Чаще всего для катионов выделяют две основные классификации: сероводородную и кислотнощелочную. Они положены в основу схем систематического анализа катионов. Сероводородная схема анализа была предложена более 100 лет назад известным русским химиком К.К.Клаусом и с некоторыми изменениями сохранилась до настоящего времени (табл.1).
Таблица1
Сероводородная классификация катионов
Группа |
|
|
|
|
|
|
Катионы |
|
|
|
|
|
Групповой |
||||||
катионов |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Реагент |
|
|
|
|
|
|
|
|
|
|
|
|||||||||
I |
K+ ,Na+ ,NH4+ ,Mg2+ |
|
|
|
|
|
|
|
|
Не имеет |
|||||||||
II |
Ba2+ ,Sr2+ ,Ca2+ |
|
|
|
|
|
|
|
|
|
|
(NH4 )2CO3 |
|||||||
III |
Fe |
3+ |
,Fe |
2+ |
,Mn |
2+ |
,Ni |
2+ |
,Co |
2+ |
,Zn |
2+ |
,Al |
3+ |
,Cr |
3+ |
(NH |
4 |
) S |
|
|
|
|
|
|
|
|
|
|
2 |
|||||||||
|
|
|
|
||||||||||||||||
IV |
Ag+ ,Hg22+ ,Pb2+ ,Cu2+ ,Cd2+ ,Bi3+ ,Hg2+ |
|
(H2S([H+ ] = 0.3 M) |
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
V |
As,Sn,Sb |
|
|
|
|
|
|
|
|
|
|
|
|
(H2S([H+ ] = 0.3 M) |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
с последующим |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
растворением |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
сульфидов в поли- |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
сульфиде аммония |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(NH4 )2S2 |
||
Систематический ход анализа начинается с осаждения суммы нерастворимых сульфидов IV и V аналитических групп действием сероводорода в кислой среде и последующем разделением смеси полученных сульфидов действием полисульфида аммония. Затем действием сульфида аммония осаждают катионы третьей аналитической группы в виде смеси сульфидов и гидроксидов. В оставшемся растворе осаждают катионы второй группы в виде карбонатов действием карбоната аммония в присутствии аммонийных солей. Оставшийся раствор содержит только катионы первой группы.
265
При изучении качественного анализа рассмотрите не только основы систематического хода анализа, но, главное, разберитесь в характерных реакциях катионов, которые и определяют построение схемы систематического разделения смеси катионов на группы, последующее разделение на подгруппы и открытие каждого катиона в отдельности с помощью специфических реакций.
Аналитическая классификация анионов может быть основана на их окис- лительно-восстановительных свойствах, на отношении к действию нитрата серебра и хлорида бария. Самостоятельно разберите эти вопросы по учебнику
[5,6].
В основном при проведении систематического качественного анализа, разделении катионов и анионов на группы используются неорганические реагенты.
Однако специфические реакции, маскирование чаще всего основаны на ис-
пользовании органических аналитических реагентов. В традиционном пред-
ставлении органические реагенты – это соединения, применяемые для обна-
ружения, отделения или количественного определения как неорганических, так и органических соединений, но прежде всего – это комплексообразую-
щие реагенты на ионы металлов. Поиск селективных органических реагентов является одним из важных направлений развития аналитической химии.
Для обнаружения и идентификации органических веществ можно использовать химические методы, но значительно информативнее результаты инструментальных методов анализа: инфракрасной спектроскопии, хромато-масс- спектрометрии.
266
Приложение 6
Методы разделения и концентрирования
Требования к пределу обнаружения примесей, начиная со второй половины XX в. повышались очень значительно и быстро в связи с развитием производства и появлением новых материалов. В начале 40-х гг. XX в. примеси в металлах определяли с ПрО до 0,1…0,01 %. К 50-м гг. эти требования возросли до 10 4 % , а для примесей в материалах для ядерной промышленности до
10 5 10 6 % (определение примесей бора, кадмия, гадолиния). С развитием полупроводниковой техники требования к чистоте полупроводниковых материалов – германия и кремния – еще ужесточились: содержание примесей никеля или меди в этих материалах недопустимо уже на уровне 10 7 10 8 %.Сле-
довательно, на 10 млрд атомов кремния или германия требовалось опреде-
лить содержание всего одного атома элемента –примеси !
Постоянно увеличивающиеся требования различных развивающихся новых отраслей промышленности к снижению ПрО дали толчок для развития аналитической химии в двух направлениях: первое - разработка новых пря-
мых методов снижения абсолютного ПрО: радиоактивационного (ПрО до
10 6 10 10 % некоторых элементов - примесей в полупроводниковых материалах), масс-спектрометрического с искровым источником ионов (возможность определения около 60 элементов с ПрО 10 5 10 6 % ). Однако высокая стоимость аппаратуры не могла обеспечить полного решения новых проблем с необходимостью снижения ПрО.
Это обстоятельство явилось толчком для развития второго направления в анализе малых концентраций – разработка приемов предварительного кон-
центрирования примесей для последующего определения их содержания в концентрате атомно-эмиссионным и другими доступными методами (например, вольтамперометрией с предварительным электролитическим накоплением и др.). Изучать эти методы мы будем позже, в следующих разделах курса, а пока нам необходимо разобраться в сути химических процессов, протекающих в процессе концентрирования, и используемых для этого химических реакциях.
267
Итак, осаждение и соосаждение как методы разделения и концентрирования применяют для разделения неорганических веществ, используя при этом неорганические и органические реагенты.
Для разделения используют осаждение в виде:
1)гидроксидов;
2)некоторых кислот (кремниевой, оловянной, вольфрамовой, танталовой, ниобиевой);
3)сульфидов;
4)сульфатов, хлоридов;
5)карбонатов, фосфатов, оксалатов;
6)осадков, образуемых с органическими реагентами.
Практически во всех названных случаях разделение может быть проведено как групповое с отделением группы элементов, образующих трудно растворимые соединения с данным групповым реагентом, либо использование того же группового реагента, но в разных дополнительных условиях (например, при разных значениях рН или в присутствии определенных комплексообразователей) позволит селективно отделить один или несколько компонентов от других.
Впервых трех случаях осаждение для разделения ионов металлов фактически используют регулирование рН. Это наиболее универсальный и легко осуществляемый способ разделения, прежде всего потому, что растворимости гидроксидов, гидратированных оксидов и кислот разных элементов существенно отличаются. Далее, концентрацию ионов водорода и гидроксида можно менять
вшироких пределах (до 1015 раз) и поддерживать с помощью буферных растворов.
Ваналитической практике используются три основных метода разделения при контролируемой кислотности:
1)разделение в растворах сильных кислот достаточно высокой концентрации;
2)разделение действием достаточно концентрированных растворов гидроксидов калия или натрия;
3)разделение в присутствии буферных растворов с промежуточными значениями рН.
Разделение с использованием органических осадителей
Органические осадители, используемые для разделения в неорганическом анализе, обладают значительно более высокой селективностью.
268
Неорганические коллекторы
Для определения содержания микроэлементов как химическими, так и физическими методами необходимо провести их предварительное концентрирование. Если для этого используют реакции осаждения, то необходимо применение коллекторов, неорганических и органических.
В качестве неорганических коллекторов могут быть использованы гидроксиды железа, алюминия, сульфиды молибдена, фосфаты и другие вещества,
образующие аморфные осадки с большой активной поверхностью. Увеличение концентрации осажденных на их поверхности микроэлементов (например, РЗЭ, Zr, As, Bi, Ga, Te,Ti, In,Se, Sb, Sn - в материалах высокой чистоты; различных минералах, горных породах и т.д.) позволяет определить содержание последних, чаще всего с помощью физических методов.
Действие неорганических коллекторов обусловлено их ионной природой и наличием поверхностных дефектов, приводящих к неравномерному распределению зарядов на поверхности.
Органические коллекторы
Более избирательными свойствами соосаждения обладают органические коллекторы. С их помощью можно селективно выделить микрокомпонент, когда его отношение к макрокомпоненту составляет 1:1015 .
В основном микроэлементы соосаждаются на поверхности индифферентных органических коллекторов, когда в раствор вводят дополнительный органический реагент, содержащий характерную аналитическую группировку на определенный микрокомпонент или группу микрокомпонентов. Например, 8- оксихинолин используют для соосаждения микроколичеств Hf и Zr или 1- нитрозо-2-нафтол - для соосаждения микроколичеств Cd, Cu, Co, U, Cr, Mn.
Соосаждение с органическими коллекторами происходит в виде хелатов или ионных ассоциатов.
Для количественной оценки методов разделения и концентрирования используются степень извлечения и коэффициент концентрирования.
Степень извлечения вещества R из фазы I в фазу II оценивается как отношение
R |
|
QI I |
|
, |
|
Q |
|
|
|||
|
|
Q |
|
||
|
II |
|
I |
|
|
где QII и QI - количество вещества во второй и в первой фазах соответственно. Для полного извлечения величина R должна быть близка к 100 % но на
практике извлечение считают количественным при условии, что R 99,9 % .
269

Коэффициент концентрирования SK
S q / qпроба ,
K Q Qпроба
где q и qпроба – количество микрокомпонента в концентрате после концентрирования и в исходной пробе; Q и Qпроба количество макрокомпонента в концентрате и в пробе.
Следовательно, величина SK позволяет оценить, во сколько раз изменилось отношение абсолютных количеств микро- и макрокомпонентов в концентрате по сравнению с тем же отношением в исходной пробе.
Как мы уже говорили, разработанные для аналитических целей методы разделения и концентрирования очень часто используются в различных областях, например при очистке различных материалов, при очистке чистых и сверхчистых материалов в производстве для электронной промышленности, полупроводниковых материалов, материалов для ракетной техники, в ядерных технологиях, при анализе конструкционных материалов, используемых для изготовления ядерных реакторов, при переработке руд черных и цветных металлов, при переработке отвалов различных месторождений в горнодобывающей промышленности для извлечения ценных микроэлементов для дальнейшего использования, при разработке методов охраны окружающей среды.
В аналитической службе предприятий машиностроения эти методы нужны для определения содержания микропримесей, когда ПрО используемого метода (например, различных видов спектроскопии) значительно превышает их содержание в анализируемом материале. Мы неоднократно будем сталкиваться с необходимостью использования разделения и концентрирования при изучении различных методов анализа чистых металлов, сплавов, композиционных материалов, используемых в машиностроительной промышленности.
270
Приложение 7
Классификация основных химических методов анализа
271
Приложение 8
Вычисления в титриметрическом анализе
Единицы количества вещества
При вычислениях в количественном анализе используются следующие термины и понятия, принятые в Международной системе единиц (СИ), которые Вы изучали в курсе неорганической химии:
Моль – количество вещества, содержащее столько определенных условных частиц, сколько атомов содержится в 0,012 кг (или в 12 г ) изотопа 12 C , то есть 6,02045 1023.
Молярная масса М – это масса 1 моль вещества.
Эквивалент – это условная частица вещества, которая в данной кислотноосновной реакции эквивалентна (равноценна) одному иону водорода или в данной окислительно-восстановительной реакции одному электрону. Эквивалент – это безразмерная величина.
Фактор эквивалентности – это число, обозначающее, какая доля реальной частицы вещества эквивалентна одному иону водорода в данной кислотноосновной реакции или одному электрону в данной окислительно-восстано- вительной реакции.
Молярная масса эквивалента вещества Х - это масса одного моль эквивалента этого вещества, равная произведению фактора эквивалентности на молярную массу М вещества
M( fýêâ (X )X ) fýêâ (X ) M(X ).
Следовательно, молярные массы эквивалентов кислот, оснований и солей зависят от стехиометрии реакций, в которых они участвуют, а в окислительновосстановительных реакциях - от числа электронов (табл. 2).
272
|
|
|
Таблица 2 |
|
Расчет молярной массы эквивалента вещества |
||
|
|
||
Реакция и фактор эквивалентности |
Молярная масса эквивалента |
||
|
|
|
|
H3PO4 + NaOH = NaH2PO4 |
+ H2O |
M (1 H3PO4 ) = |
|
|
f'экв(H3PO4 ) = 1 |
|
= fэкв(H3PO4 ) M (H3PO4 ) = |
|
|
|
= 1 98 г/моль= 98 г/моль |
|
|
|
|
H3PO4 |
+ 2NaOH = Na2HPO4 |
+ 2H2O |
M(1/2 H3PO4) = |
|
fэкв(H3PO4 ) = 1/2 |
|
= fэкв(H3PO4) M(H3PO4) = |
|
|
|
= (1/2) 98 г/моль= 49 г/моль |
|
|
|
|
H3PO4 |
+ 3NaOH = Na3PO4 |
+ 3H2O |
M(1/3 H3PO4) = |
|
fэкв(H3PO4 ) = 1/3 |
|
= fэкв(H3PO4) M(H3PO4) = |
|
|
|
= (1/3) 98 г/моль= 32,66 г/моль |
|
|
|
|
Ca(OH) |
2 + HCl = Ca(OH)Cl + H2O |
M (1 Ca(OH)2 ) = |
|
|
fэкв (Ca(OH)2 ) = 1 |
|
= fэкв (Ca(OH)2 ) M (Ca(OH)2 )= |
|
|
|
= 1 74,08 г/моль = 74,08 г/моль |
|
|
||
Ca(OH)2 + 2HCl = CaCl2 + 2H 2O |
M (1/2 Ca(OH)2 ) = |
||
fэкв (Ca(OH)2 ) = 1/2 |
|
= fэкв(Ca(OH)2 ) M (Ca(OH)2 ) = |
|
|
|
|
= (1/2) 74,08 г/моль = 37,04 г/моль |
|
|
|
|
Na2CO3 |
+ 2HCl = 2NaCl + H2O + CO2 |
M (1/2 Na 2CO3 ) = |
|
|
fэкв (Na2CO3 ) = 1/2 |
= fэкв (Na 2CO3 ) M (Na 2CO 3 ) = |
|
|
|
|
= (1/2) 106 г/моль = 53 г/моль |
|
|
||
Na2CO3 + HCl = NaHCO3 + NaCl |
M (Na 2 C O 3 ) = |
||
|
fэкв(Na2CO3 ) = 1 |
|
= fэкв (N a 2 CO 3 ) M (Na 2 C O 3 ) = |
|
|
|
= 1 106 г/моль = 106 г/моль |
|
|
|
|
273
Способы выражения концентрации растворов
Способы выражения концентрации растворов Вы также изучали в курсе неорганической химии, за исключением специфических для количественного анализа способов выражения концентрации (титр) и размерностей, используемых при выражении концентрации в долях.
Единицей объема в количественном анализе служит кубический дециметр (дм3), который в точности равен 1литру (л); соответствующие кратные единицы
– см3 и мл.
Молярная концентрация с – это отношение количества моль n растворенного вещества S к объему раствора V
c(S) n(S) /V моль/дм3 или моль/л.
Молярная концентрация эквивалента – это число моль эквивалентов вещества, растворенных в 1 литре раствора. Например, 0,1 н Na2CO3 (fэкв = 1/2) означает ту же концентрацию, что и 0.1 М (1/2 Na2CO3) или 0,1 N Na2CO3 .
С целью окончательного уяснения разницы в понятиях и способах записи молярной концентрации и молярной концентрации эквивалента сопоставьте рассмотренные значения концентраций с соответствующим каждому из них значению массового содержания вещества в растворе ( табл. 3).
Таблица 3
Способы выражения концентраций растворов и соответствующие им массовые содержания веществ в растворах
Способ выражения |
|
Массовое содержание |
концентрации |
|
вещества |
|
|
|
с (HCl) = 1 моль/л |
36,5 |
г/л HCl |
|
|
|
с (H2SO4) = 1 моль/л |
98,0 |
г/л H2SO4 |
с (1/2 H2SO4) = 1 моль/л |
49,0 |
г/л H2SO4 |
с (KMnO4) = 1 моль/л |
158,0 г/л KMnO4 |
|
с (1/5 KMnO4) = 1 моль/л |
31,6 |
г/л KMnO4 |
с (1/3 KMnO4) = 1 моль/л |
52,6 |
г/л KMnO4 |
Массовая концентрация (символ , единица – г/л) равна массе растворенного вещества S (m(S)), деленной на объем раствора:
(S) m(S) /V .
274
Если для массовой концентрации пользуются кратной единицей г/мл, то массовую концентрацию называют титром.
Титр стандартного раствора вещества S (символ Т(S), единица измере-
ния – г/мл) – это концентрация стандартного раствора, равная массе вещества S (m(S)), содержащегося в 1 мл раствора:
T(S) m(S) /V .
Например, Т (НСl) = 0,01 г/мл означает, что в 1 мл стандартного раствора HCl содержится 0,01 г хлороводорода.
При серийных определениях удобно пользоваться для проведения расчетов понятием титра по определяемому веществу.
Титррастворапоопределяемомувеществу– этомассаопределяемоговещест-
ва, эквивалентная 1 мл стандартного раствора. Например, Т(HCl/NaOH) = 0,01 г/мл означает, что0,01 гNaOH эквивалентен1 млстандартногораствораHCl.
Массовая доля растворенного вещества S (символ (S), безразмерная величина, вычисляется в долях от единицы или в процентах) – равна отношению массы растворенного вещества m (S) к массе раствора mp
(S) m(S) / mp .
Если известны плотность раствора и объем раствора Vр, то
(S) m(S) /( Vp ) .
Запись (HCl) = 0,2 обозначает 20 %-ный раствор хлороводорода, или что массовая доля хлороводорода в растворе равна 20%. Употребление терминов «процентная концентрация», «весовая часть» не рекомендуется.
Понятие доли компонента используется также и при анализе твердых и газообразных веществ. В этом случае независимо от агрегатного состояния анализируемого объекта доля компонента показывает отношение числа частей компонента к общему числу частей объекта. Число частей может быть выражено в разных единицах, в зависимости от которых различают молярную , мас-
совую , объемную доли:
α(ni / n),
ω(mi / m),
(Vi / V ).
Долю можно выразить различными способами. Для этого значения долей умножают на 10n, где n = 2, 3, 6, 9, причем:
если множитель n равен 102, то доля выражается в процентах;
275

если множитель n равен 103, то доля выражается в промилях ppt (parts per thousand или число частей на тысячу частей объекта);
если множитель n равен 106, то доля выражается в миллионных долях ppm (parts per million или число частей на миллион частей объекта);
если множитель n равен 109, то доля выражена в миллиардных долях объекта ppb (parts per billion или число частей на биллион частей объекта).
Стандартные растворы
Стандартным раствором называется титрант с точно известной концентрацией. Точность приготовления раствора определяется точностью, предъявляемой к результатам анализа.
По способу приготовления различают первичные и вторичные стандарт-
ные растворы.
Первичные стандартные растворы готовят растворением навески хими-
чески чистого вещества, состав которого точно соответствует его стехиометрии, в мерной колбе (рис.1).
Рис. 1. Мерные колбы Например, в методах окислительно-восстановительного титрования ис-
пользуются: дихромат калия, щавелевая кислота или оксалат натрия, сублимированный иод; в комплексонометрии – это динатриевая соль этилендиаминтетрауксусной кислоты. Но таких веществ очень немного. Так, растворы кислот или оснований для кислотно-основного титрования можно приготовить только
276
приблизительной концентрации путем разбавления концентрированных растворов.
Вторичные стандарты или стандартизированные растворы получают после их стандартизации с помощью установочных веществ (первичных
стандартов).
Установочные вещества (первичные стандарты) подбираются в зависимости от типа рассматриваемого взаимодействия:
кислотно-основное титрование,
окислительно-восстановительное титрование,
осадительное титрование,
комплексометрическое титрование.
Установочное вещество может быть взято
в виде навески,
в виде точного объема раствора известной концентрации.
При анализе сложных объектов в качестве первичных стандартов могут быть использованы навески стандартных образцов, аналогичных по составу анализируемому объекту.
С примерами определения Вы ознакомитесь при выполнении контрольной задачи №2 и при работе в лаборатории (лабораторные работы №1 и №2).
277
Приложение 9
Метрологические основы аналитической химии
При обнаружении компонента, то есть проведении качественного анализа или идентификации компонента, фиксируют появление аналитического сигнала, например появление окраски, осадка в растворе и т.д.
При определении количества компонента, то есть при проведении коли-
чественного анализа, на заключительной стадии анализа измеряют значение ве-
личины аналитического сигнала, например замеряют массу осадка в грави-
метрии, объем раствора в титриметрии, в физико – химических методах анализа – интенсивность какого-либо физического сигнала ( интенсивность спектральной линии, величину потенциала, силу тока и т. д.).
Аналитический сигнал представляет собой среднее из измерений какойлибо физической величины S, функционально связанной с содержанием с определяемого компонента соотношением
S f (c) .
Химический анализ сложных объектов включает много разнообразных операций, каждая из которых даже при самом тщательном выполнении приво-
дит к появлению погрешности, то есть отклонению результата от истин-
ного значения.
Поэтому по окончании анализа необходимо рассчитать погрешность оп-
ределения, которая является одной из метрологических характеристик методики анализа.
Для этого необходимо усвоить ряд понятий, которые используются для оценки полученных результатов анализа и называются метрологическими ха-
рактеристиками аналитических методик.
Метрология – это наука об измерениях, методах достижения их единства и требуемой точности измерений (от греч. metron - мера, logos – слово, учение).
Метрологические характеристики метода включают:
погрешность измерения;
правильность;
воспроизводимость;
интервал определяемых содержаний;
чувствительность определения, характеризуемую пределом определения
ПрО.
278
Оценка достоверности полученных результатов анализа проводится на основе вычисления погрешности химического анализа.
Погрешностью измерения называется отклонение результата измерения от истинного значения измеряемой величины.
Классификация погрешностей проводится по нескольким разным призна-
кам:
по способу вычисления (абсолютная и относительная погрешности);
по характеру вызывающих их причин (систематические и случайные);
по способу обработки результатов параллельных определений (средние арифметические и среднеквадратичные).
Абсолютная погрешность единичного определения ∆xi равна разности
между результатом единичного определения xi и истинным значением этой величины .
Но чаще определяют абсолютную погрешность анализа ∆, равную разности между средним измерением величины x и истинным значением этой величины μ.
xi xi μ,
x μ.
Абсолютная погрешность может быть положительной или отрицатель-
ной величиной в зависимости от того, как она влияет на результаты анализа: за-
вышает или занижает их.
Относительная погрешность измерения D это отношение абсолютной погрешности к истинному значению измеряемой величины.
Эта величина выражается в долях или процентах и знака не имеет
D / μ èëè D / μ) 100 %.
Впрактике анализа истинное значение содержания анализируемого компонента в пробе остается неизвестным и вместо него используют среднее арифметическое значение, полученное из нескольких параллельных определений.
Входе выполнения лабораторных работ Вы научитесь рассчитывать эти величины для оценки получаемых результатов.
По характеру вызывающих причин погрешности могут быть систематиче-
скими и случайными.
279
Систематическая погрешность остается постоянной при повторных измерениях или меняется по одной закономерности.
Ее значение или завышает, или занижает результат, то есть ее знак остается постоянным во всех измерениях. Следовательно, систематическая погрешность вызвана постоянно действующей причиной: например, инструментальные погрешности, связанные с используемой для измерения аналитического сигнала аппаратурой (некалиброванная мерная посуда, непроверенные разновесы - в химических методах анализа; в инструментальных методах – невысокий класс точности измерительного прибора, износ, старение, разрегулировка прибора), методические погрешности, обусловленные методикой определения.
Случайная погрешность при повторных измерениях изменяется случайным образом.
Числовое значение и знак случайной погрешности меняется, причины их возникновения неизвестны; их оценка производится методами математиче-
ской статистики.
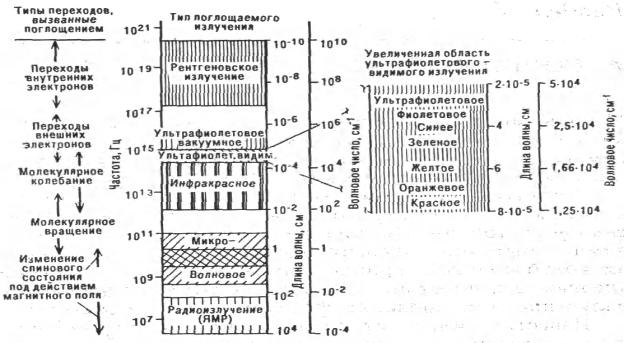
Приложение 10
Электромагнитный спектр излучения
280
Приложение 11
Основные характеристики электромагнитного излучения
Частота колебаний ν показывает число колебаний в 1 с; единицей измерения является герц (Гц).
Длина волны λ показывает наименьшее расстояние между точками, колеблющимися в одинаковых фазах; измеряется в метрах (м) и его долях, имеющих разную размерность в разных областях спектра: см (10-2 м) и мкм (10 -6 м)– в инфракрасной области спектра, нм (10 -9 м) – в видимой и ультрафиолетовой (см. Приложение 10).
В вакууме (и приближенно в воздухе) длина волны и частота колебаний связаны между собой соотношением
ν c / λ |
|
|
где с – скорость распространения света в вакууме 2,998·10 8 м·с-1. |
|
|
В инфракрасной спектроскопии часто |
используется волновое число |
|
|
-1 |
). |
ν 1/ λ. Эту величину принято выражать в обратных сантиметрах (см |
||
Из Приложения 10 мы видим, что для разграничения отдельных областей спектра могут быть использованы различные единицы измерения длин волн и частоты. Частота ν и длина волны λ электромагнитного излучения связаны между собой соотношением c λ ν, где с − скорость распространения электромагнитных волн, равная в вакууме с = 3 · 1010 cм · с-1. В воздухе она на 0,03 % меньше. Частота излучения при переходе из одной среды в другую не меняется. Следовательно, меняется длина волны. Этим изменением (на 0,03 %) обычно пренебрегают.
Вобласти радио- и микроволн в качестве единиц измерения частоты ν используют герцы, килогерцы и мегагерцы (1 Гц = с-1 − одно колебание в течение секунды).
Винфракрасной области (при частотах выше 1012 Гц) точность измерения частот по сравнению с точностью измерения длин волн уже неудовлетворительна. Поэтому в качестве единицы измерения используют сантиметр или дольные единицы от него. Кроме того, вместо длины волны используется зна-
чение ее обратной величины 1/ , называемое волновым числом и обозначаемое ν. Эта величина показывает число волн света, помещающихся в той единице измерения длины, которая использована для выражения длины волны .
281
Поэтому в инфракрасной (ИК) области волновое число выражают в обратных сантиметрах (см-1), то есть указывают число волн, помещающихся в одном сантиметре.
В ультрафиолетовой (УФ) и видимой (В) областях спектра длину волны излучения обычно выражают в нанометрах (1нм = 10-9 м).
282
Приложение 12
Методы атомной спектроскопии
Атомная спектроскопия основана на поглощении или испускании рентге-
новского, видимого или УФ-излучения. Она используется для качественного и количественного анализа.
Рентгеновское излучение и поглощение обусловлено возбуждением внутренних, не участвующих в образовании связей электронов. Поэтому характер
рентгеновского атомного спектра не зависит от химического состояния элемента в образце, а рентгеновский анализ проводится без разрушения образца.
Ввидимой и УФ-области поглощение или излучение обусловлено возбуждением внешних валентных электронов. Поэтому чистый атомный спектр
можно получить только после отделения анализируемого элемента от других связанных с ним элементов. С этой целью проводят атомизацию образца, в процессе которой молекулы распадаются на составные части и превращаются в атомы и ионы, существующие в газообразном состоянии.
Атомные спектры и поглощения, и испускания имеют дискретный характер (линейчатая структура). Длины волн такого спектра характерны только для данного элемента, они носят только сугубо индивидуальный характер. Спектры некоторых атомов, например натрия или лития, просты и состоят из очень небольшого числа линий. Наоборот, в спектрах d- и f-элементов, например железа, насчитываются десятки тысяч спектральных линий, которые отчетливо воспроизводятся. На этом основан элементный качественный анализ. Зависимость между интенсивностью отдельных линий и количеством элемента в пробе лежит в основе количественного анализа.
Ватомной спектроскопии используется целый ряд способов атомизации анализируемого образца. Пламя, дуга и искра являются традиционными. Методы, основанные на их использовании, хорошо методически отработаны. В последние годы в связи с появлением новых задач появился целый ряд новых источников, таких как источник индуктивно-связанной плазмы, микроволновый разряд, лазерные атомизаторы (табл. 4).
Подробно этот материал будет рассмотрен в соответствующих разделах.
283
Таблица 4
Классификация методов атомной спектроскопии |
|
|
|||||
Название |
Метод |
Источник |
Способ введения |
|
|||
Метода |
атомизации |
Излучения |
|
пробы |
|
|
|
|
Абсорбционные методы |
|
|
|
|
||
Пламенная атом- |
Пламя |
Лампа с |
полым |
Анализируемый |
рас- |
||
но- |
|
катодом |
|
твор |
распыляют |
в |
|
абсорбционная |
|
|
|
пламени |
|
|
|
Непламенная |
Нагретая поверх- |
То же |
|
Анализируемым |
рас- |
||
атомно- |
ность |
|
|
твором капают на на- |
|||
абсорбционная |
|
|
|
гретую поверхность |
|
||
Рентгеноабсорб- |
Не требуется |
Рентгеновская |
Определяемое веще- |
||||
ционная |
|
трубка |
|
ство помещают в по- |
|||
|
|
|
|
ток излучения |
|
|
|
|
|
|
|
|
|
|
|
|
Эмиссионные методы |
|
|
|
|
||
Пламенно- |
Пламя |
Пламя |
|
Анализируемый |
рас- |
||
эмиссионная |
|
|
|
твор |
распыляют |
в |
|
|
|
|
|
пламени |
|
|
|
|
|
|
|
|
|||
Дуговая |
Электрическая |
Дуга |
|
Определяемое веще- |
|||
|
Дуга |
|
|
ство помещают в по- |
|||
|
|
|
|
лость электрода |
|
|
|
Искровая |
Электрическая |
Искра |
|
То же |
|
|
|
|
Искра |
|
|
|
|
|
|
Спектроскопия с |
Электрогенери- |
ИСП |
|
Анализируемый |
рас- |
||
индуктивно свя- |
рованная плазма |
|
|
твор |
распыляют |
в |
|
занной плазмой |
в газе − носителе |
|
|
виде аэрозоля в газ − |
|||
|
(индуктивно- |
|
|
носитель |
|
|
|
|
связанная плазма |
|
|
|
|
|
|
|
ИСП, ICP) |
|
|
|
|
|
|
Атомно-флу- |
Пламя; плазма |
Разрядная |
лам- |
Анализируемый |
рас- |
||
оресцентная |
|
па; лазер |
|
твор |
распыляют |
в |
|
|
|
|
|
пламени или плазме |
|
||
Рентгенофлуо- |
Не требуется |
Рентгеновская |
Определяемое веще- |
||||
ресцентная |
|
трубка |
|
ство |
помещают |
на |
|
|
|
|
|
пути |
рентгеновских |
||
|
|
|
|
лучей |
|
|
|
284
Приложение 13
Фурье-спектрометры
В недиспергирующих спектрометрах (фурье–спектрометры) вместо монохроматора применяют интерферометры. Регистрируемая детектором интерферограмма содержит информацию об изменении интенсивности каждой частоты в спектре источника, которое вызвано поглощением образца. Регистрация всего спектра фурье-спектрометром занимает несколько секунд, а для записи ИКспектра сканирующим спектрофотометром требуется несколько минут. Интерферограмма по специальной программе обрабатывается компьютером и преобразуется в обычный ИК-спектр. Преимущества фурье–спектрометров перед классическими дисперсионными щелевыми заключаются в большей светосиле, большей разрешающей способности и возможности одновременного измерения всех компонент спектра. Особенно это важно для исследования протяженных спектров слабых поглощений в далекой ИКобласти (400…10 cì -1 ).
В настоящее время на рынке аппаратуры для ИКобласти достаточно широко представлены различные модели фурье-спектрометров (Приложение
15…16).
ИК-спектроскопия и, в частности фурье-спектрометры используются в аналитической службе машиностроительной отрасли как один из вариантов идентификации органических соединений.
285
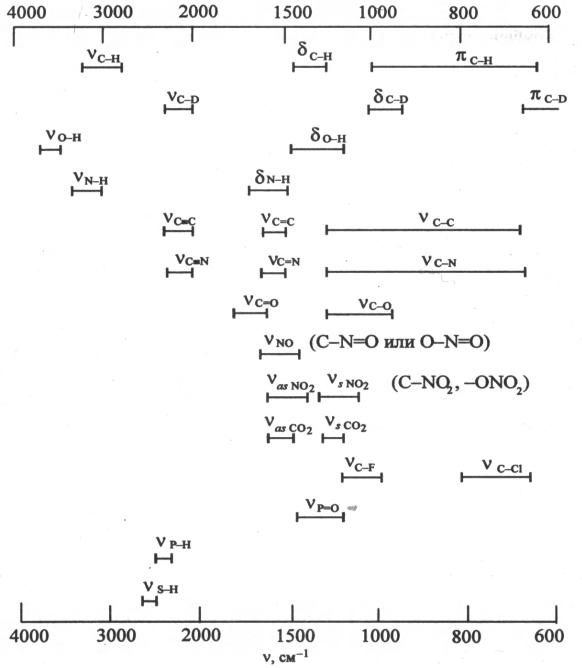
Приложение 14
Характеристические частоты некоторых функциональных групп в органических соединениях
286
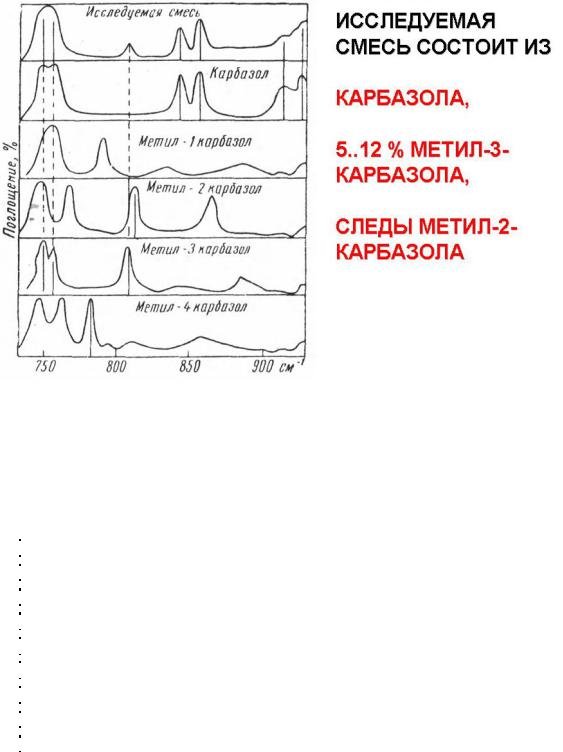
Приложение 15
Пример структурного анализа органических веществ в ИК-области
Приложение 16
Основные фирмы, представленные на современном рынке аналитического приборостроения
 PERKIN - ELMER,
PERKIN - ELMER,
 VARIAN,
VARIAN,
 HEWLETT PACKARD,
HEWLETT PACKARD,
 HITACHI,
HITACHI,
 SHIMADZU,
SHIMADZU,
 INSTRUMENTATION LABORATORY,
INSTRUMENTATION LABORATORY,
 THERMO SPECTRONIC (USA),
THERMO SPECTRONIC (USA),
 PYE-UNICAM,
PYE-UNICAM,
 THERMO ELECTRON CORPORATION
THERMO ELECTRON CORPORATION
287

Приложение 17
Образцы продукции аналитического приборостроения. Спектрофотометры
288

Образцы продукции аналитического приборостроения
289
Приложение18
Метод атомно-абсорбционной спектроскопии
Теоретические основы метода
Атомный пар получают распылением раствора анализируемого образца в пламени. Раствор подается в пламя горелки в виде аэрозоля. Растворитель испаряется, удаляется кристаллизационная вода, большая часть компонентов металлического характера восстанавливается до элементного состояния и при этом находится в пламени в основном невозбужденном состоянии.
Если на эти атомы направить электромагнитное излучение с энергией, точно соответствующей энергии характеристических электронных переходов, то произойдет поглощение.
Атомное поглощение точно так же, как и молекулярное, характеризуется экспоненциальным законом убывания интенсивности проходящего светового потока в зависимости от длины поглощающего слоя (толщины слоя плазмы), концентрации поглощающих атомов.
Величина оптической плотности атомного пара (А) в соответствии с основным законом светопоглощения прямо пропоциональна концентрации поглощающих частиц (ñàò .) - атомов определяемого элемента в атомизаторе:
A kàò lcàò . ,
где kàò - коэффициент поглощения света свободными атомами; l - длина оптического пути (толщина светопоглощающего слоя – пламени). Постоянство величины l достигается с помощью горелок специальной конструкции.
Эта зависимость аналогична закону Бугера-Ламберта-Бера и графически выражается прямой линией, но коэффициенты атомного поглощения примерно на три порядка выше по сравнению с молярными коэффициентами поглощения для цветных реакций в водных растворах (примерно n·108 и n·105 соответственно).
Методом атомной абсорбции определяются трудно возбуждающиеся элементы. Наибольшую чувствительность метод атомно-абсорбционной спектроскопии (ААС) проявляет при определении As, Be, Cd, Bi, Hg, Mg, Pb, Te, Zn, Cs, In.
Элементы с низкими потенциалами возбуждения, такие как Li, K, Na, Ba, Sr, Tl, легко возбуждаются в пламени, и потому для их определения значительно более чувствительным оказывается пламенно-эмиссионный метод.
290
Молекулы вещества поглощают свет полосами в широких интервалах длин волн, а поглощение парами атомов происходит в узких пределах, порядка тысячных долей нанометра. Атомный пар поглощает электромагнитное излучение с энергией, точно соответствующей энергии характеристических электронных переходов. Поскольку условия атомизации подобраны таким образом, что основная часть атомов находится в невозбужденном состоянии, то пламен-
ные атомно-абсорбционные спектры состоят преимущественно из резонансных линий, отвечающих переходам из основного в возбужденное состояние, а
линии, связанные с переходами из одного возбужденного состояния в другое, оказываются слишком слабыми, чтобы их можно было обнаружить.
В связи с тем, что во-первых, атомно-абсорбционные линии очень узки, а во-вторых, каждый элемент обладает характерным только для него, собственным, набором разрешенных энергетических переходов, то аналитический метод, основанный на использовании атомной абсорбции, должен обладать очень высокой селективностью.
Но одновременно с возможностью реализации в этом методе столь важного для решения аналитических задач фактора селективности узость атомноабсорбционной линии вызывает и необходимость решения новой, по сравнению с методом молекулярной абсорбции, задачи. Дело в том, что линейное соотношение между оптической плотностью и концентрацией наблюдается при условии, что ширина полосы мала по сравнению с шириной линии в максимуме. Но современные монохроматоры не способны выделить полосу излучения, сопоставимую по ширине с атомно-абсорбционной линией (0,002…0,005 нм).
Поэтому если для атомной абсорбции использовать непрерывный источник с монохроматором, то лишь незначительная часть излучения будет иметь нужную длину волны, и относительное изменение интенсивности возникающей полосы будет мало по сравнению с изменением излучения в максимуме. В этом случае нарушается линейная зависимость между величиной абсорбции и концентрацией, а чувствительность метода снижается.
Поэтому в качестве источников излучения в атомно-абсорбционных спектрометрах должны быть использованы особые источники излучения, которые испускают линии значительно более узкие, чем абсорбционные линии.
Этому требованию отвечают разрядные лампы – лампы с полым катодом и безэлектродные разрядные лампы, которые являются источниками линейчатых спектров.
291

Принципиальная схема атомно-абсорбционного спектрометра
Принципиальная схема установки атомно-абсорбционного спектрометра представлена на рис.2
Рис. 2. Принципиальная схема установки атомно-абсорбционного спектрометра
1 – лампа с полым катодом; 2 – пламя; 3 - горелка–атомизатор; 4 – устройство для измерения и регистрации сигнала
Таким образом источником характеристического излучения атомов исследуемого элемента является лампа с полым катодом, который изготавливается из определяемого металла или покрывается слоем этого металла. Анод изготавливается из вольфрама. Если к электродам приложить напряжение, газ ионизируется, ионы мигрируют к электродам. Некоторые из этих атомов возбуждаются и испускают резонансное излучение, которое может быть поглощено только невозбужденными атомами определяемого элемента.
Для отделения линии, интенсивность которой необходимо измерить, от других линий источника света в схеме атомно-абсорбционного спектрометра используется монохроматор, помещенный между пламенем и детектором. В
качестве монохроматора чаще всего используют дифракционную решетку. Схема процесса резонансного поглощения в атомно-абсорбционной спек-
троскопии представлена в Приложении 19.
Безэлектродные лампы аналогичны по действию лампам с полым катодом. Они содержат небольшое количество чистого вещества или его летучего легкодиссоциирующего соединения, которое под действием микроволнового
292
поля переводится в атомный пар и возбуждается. Эти лампы используются для определения неметаллов ( As, Se, Te, P) и летучих металлов ( Hg, Rb, Cs) .
Основной недостаток разрядных ламп – это возможность определения только одного элемента. Правда, одновременно это обеспечивает и высокую селективность определения и исключает необходимость удаления или маскирования сопутствующих элементов, как в фотометрии.
В последнее время ведутся интенсивные работы по созданию ламп с перестраиваемой частотой, но пока традиционные лампы с полым катодом занимают лидирующее положение.
Поглощение и излучение фона может приводить к появлению спектральных помех, искажающих результаты, и потому требующих коррекции измерения сигнала.
Более детально разобраться в этом материале Вы можете, ознакомившись с материалами мультимедиа по теме «Атомно-абсорбционная спектроскопия» во время работы в лаборатории.
Основные типы атомизаторов в атомно-абсорбционной спектроскопии
Основным типом атомизатора до настоящего времени является пламенный, типа горелки-распылителя с предварительным смешиванием газов. При этом пламя выполняет не только функцию атомизатора, но и кюветы. Поскольку атомно-абсорбционная спектроскопия подчиняется закону Бугера- Ламберта-Бера, то чувствительность метода должна возрастать с увеличением толщины поглощающего слоя, в данном случае с увеличением длины пламени, просвечиваемом источником света. Это достигается применением специальных щелевых горелок, обеспечивающих увеличение толщины поглощающего слоя до 10 см.
В пламени анализируются все элементы, определяемые методом ААС (Приложение 21).
Пламя обеспечивает наибольшую точность измерения (погрешность менее 1 %), производительность – до 200 анализов в час. Пламенный атомизатор наиболее отработан как конструктивно, так и методически (процесс измерения и наличие методик предварительной обработки проб конкретных образцов).
Чаще всего в качестве горючего газа используется ацетилен в смеси воздухом. При этом обеспечивается температура 2300 °С , достаточная для атомизации более чем 30 элементов.
При определении редкоземельных элементов, алюминия, бериллия, вана-
293
дия, вольфрама и некоторых других, образующих труднолетучие оксиды, температура, которую обеспечивает воздушно-ацетиленовое пламя, оказывается недостаточной для их атомизации. Поэтому нужную концентрацию атомов этих элементов в пламени можно получить, только подняв его температуру до 3000 °С использованием смеси закись азота - ацетилен. Но этот вариант значительно удорожает анализ.
Наряду с использованием пламенных атомизаторов в настоящее время разработано несколько типов электротермических анализаторов (ЭТА), наиболее удачным из которых считается предложенная в 1961 г. так называемая кювета Львова. (Подробно в материалах мультимедиа.) Устройство состоит из небольшой графитовой трубки, нагреваемой при пропускании через нее тока силой до 50 А при низком напряжении. Трубка на время измерений заполняется инертным газом для предотвращения окисления поверхности графита. При нагревании в дуге постоянного тока образец полностью испаряется. Тенденция к образованию термостойких оксидов устраняется использованием восстановительной атмосферы.
Позднее были разработаны многочисленные варианты электротермических атомизаторов самых разных конструкций в виде графитовых печей, графитовых стержней, тиглей, лодочек, лент и проволочек из тугоплавких металлов (тантал, платина, вольфрам), нагреваемых током.
Преимущество непламенных атомизаторов заключается в необычайно высокой чувствительности при очень небольшом объеме пробы (0,5…10 мкл). Предел обнаружения лежит в интервале 10-10 – 10-13 г определяемого элемента. Относительная воспроизводимость непламенных методов обычно в пределах 5…10 %; при пламенной атомизации - воспроизводимость порядка 1…2 %.
Графитовые электротермические анализаторы поставляются практически всеми фирмами-производителями аппаратуры для ААС. Во всех вариантах конструкций анализируемый раствор с помощью пипетки-дозатора вводят в графитовую трубку (кювету). Время и температура нагрева регулируются по специальной программе в зависимости от методики (Приложение 20). На первой стадии печь нагревается до температуры, обеспечивающей испарение растворителя и кристаллизационной воды (100-120 °С). На второй стадии происходит озоление и разрушение солей металлов с неорганическими и органическими анионами. На третьей стадии температура повышается до 2000-3000 °С. Проба атомизируется за несколько секунд. Излучение от источника проходит непосредственно над нагретой поверхностью. В начале третьей стадии нагрева оптиче-
294
ская плотность при длине волны поглощения за несколько секунд возрастает до максимума, а затем падает до нуля, поскольку в четвертой высокотемпературной стадии печь очищается от остатков пробы путем выноса их инертным газом. После этого прибор готов для проведения анализа следующей пробы.
Ограничением для этого варианта является невозможность определения элементов, образующих труднодиссоциирующие карбиды.
Сравнительные метрологические характеристики атомизаторов представлены в Приложении 21.
Использование метода для количественного анализа
Метод ААС может быть использован только для количественного элементного анализа практически любых органических и неорганических объектов. Для определения концентрации используется метод калибровочного графика и метод добавок. Методики определения разработаны более чем для 70 элементов.
Особенно важно определение содержания примесей в различных металлах на уровне менее 10-4 – 10-5 %. Например, в золоте определяют содержание серебра, свинца, цинка, меди; в циркониипримеси кадмия и свинца. Методом ААС производится анализ загрязнений почвы тяжелыми металлами, определяется содержание металлов в удобрениях, растениях, при проведении биологических и клинических исследований.
Метрологические характеристики метода
Сравнительные возможности метода при использовании обоих вариантов атомизации приведены в Приложении 21: при использовании пламенного источника атомизации для большинства элементов пределы обнаружения находятся в интервале 1 - 30 мкг/мл, а при использовании электротермических атомизаторов (ЭТА) составляют от 0,00005 до 0,01 мкг/мл, а абсолютные значения пределов обнаружения составляют от 10-11 до 10-14 г.
Относительная погрешность результатов при использовании метода ААС не превышает 1 − 4 %.
Аппаратура ААС представлена на современном рынке аналитического приборостроения многими зарубежными фирмами (Приложение 22). Отечественные атомно-абсорбционные спектрометры (Приложение 23) в настоящее время по большинству параметров не уступают зарубежным.
295
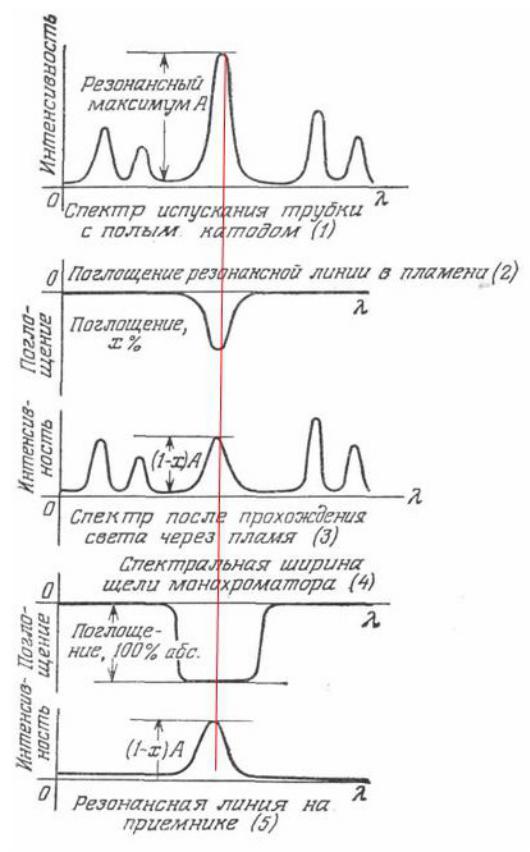
Приложение 19
Резонансное поглощение в атомно-абсорбционной спектроскопии
При- |
ложе |
же- |
ние |
20 |
|
|
Тем- |
пера |
ра- |
тур- |
ная |
про- |
|
296
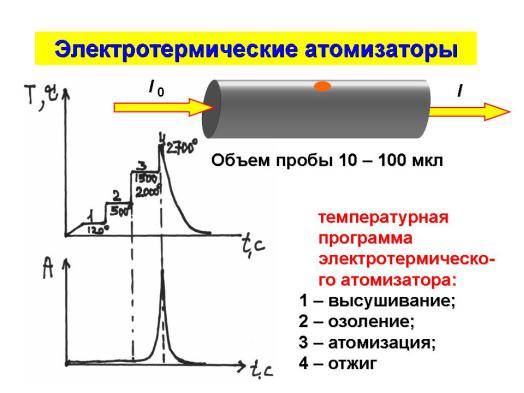
грамма электротермического атомизатора
297

Приложение 21
Пределы обнаружения элементов Сmin при атомизации пробы в пламени и электротермической атомизации (ЭТА)
Элемент |
|
ЭТА |
Пламя |
|
Сmin, г |
|
Сmin,мкг/мл |
Сmin, мкг/мл |
|
|
|
|
|
30 |
Al |
2·10-12 |
|
0,005 |
|
As |
1·10-10 |
|
0,02 |
100 |
Ca |
3·10-13 |
|
0,02 |
1 |
Cd |
1·10-13 |
|
0,0001 |
1 |
Cr |
1·10-11 |
|
0,01 |
3 |
Cu |
2·10-12 |
|
0,002 |
2 |
Fe |
3·10-12 |
|
0,005 |
5 |
Hg |
6·10-11 |
|
0,1 |
500 |
Mg |
6·10-14 |
|
0,00002 |
0,1 |
Mn |
2·10-13 |
|
0,0002 |
2 |
Mo |
1·10-11 |
|
0,005 |
30 |
Na |
2·10-13 |
|
0,0002 |
2 |
Ni |
1·10-11 |
|
0,02 |
5 |
Pb |
2·10-12 |
|
0,002 |
10 |
Sn |
5·10-12 |
|
0,1 |
20 |
B |
7·10-11 |
|
0,1 |
20 |
Zn |
5·10-14 |
|
0,00005 |
2 |
298

Приложение 22
Атомно-абсорбционные спектрометры, представленные на современном рынке аналитического приборостроения
 PERKIN - ELMER,
PERKIN - ELMER,
 VARIAN,
VARIAN,
 HEWLETT PACKARD,
HEWLETT PACKARD,
 HITACHI,
HITACHI,
 SHIMADZU,
SHIMADZU,
 INSTRUMENTATION LABORATORY,
INSTRUMENTATION LABORATORY,
 PYE-UNICAM,
PYE-UNICAM,
 THERMO ELECTRON CORPORATION
THERMO ELECTRON CORPORATION
Приложение 23
Внешний вид отечественного атомно-абсорбционного спектрометра МГА-915
299
Приложение 24
Количественные характеристики спектрального прибора
1.Угловая и линейная дисперсия.
2.Разрешающая способность.
3.Светосила.
Угловая и линейная дисперсия Угловая дисперсия определяется отношением углового расстояния между
двумя лучами к разности длин волн этих лучей.
Линейная дисперсия представляет собой отношение линейного расстояния в мм между двумя спектральными линиями в спектре к разности их длин волн.
Обратная линейная дисперсия d /dl определяется интервалом длин волн спектра, который приходится на 1 мм длины спектра (нм/мм).
Приложение 25
Разрешающая способность
Разрешающая способность R это отношение средней длины волны двух спектральных линий, которые могут быть разрешены данным спектральным прибором, к разности длин волн этих линий R = /d , фактически это возможность различить две близкие линии. Например, рассмотрим такую возможность для двух линий спектра железа FeI: 1 = 415,48 нм и 2 = 415,45 нм.
R = /d = 415,4/0,03 = 13 846.
Теоретическая разрешающая способность для однопризменного спектрального прибора находится в пределах от 5000 в длинноволновой части до 60000 в коротковолновой части спектра, а для дифракционной решетки (600 штрихов/мм) по всему спектру R = 90000.
300
Приложение 26
Светосила прибора Светосила прибора определяется величиной лучистого потока, прошед-
шего через прибор и достигшего поверхности спектра. При визуальной регистрации спектра освещенность создается на сетчатке глаза наблюдателя. При фотографической регистрации спектра лучистый поток создает освещенность на фотопластинке. При фотоэлектрической регистрации имеет значение весь лучистый поток, достигающий фотокатода приемника.
Приложение 27
|
Возбуждение излучения |
|
|
|
|
|
Источник возбуждения (атомизаторы) |
|
|
||
|
|
|
|
|
|
Пламя |
|
1500 |
|
2200 |
|
|
|
|
C |
||
Дуга |
|
3000 |
|
7000 |
|
|
|
|
C |
||
Искра |
|
7000 |
|
10000 |
|
|
|
|
C |
||
Индуктивно связанная плазма ИСП |
7000 |
|
10000 |
|
|
|
|
|
C |
||
(ICP) |
|
|
|
|
|
|
|
|
|
|
|
301
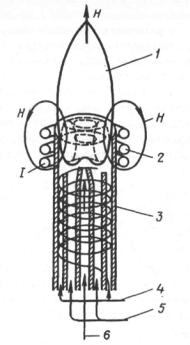
Приложение 28
Индуктивно связанная плазма (ИСП, ICP)
Схема атомизатора с ИСП
1 – зона наблюдения;
2 – индукционная катушка;
3 – кварцевая горелка;
4 – поток охлаждающего газа;
5 – промежуточный поток; 6 – анализируемый раствор в виде аэрозоля в смеси с газом (аргоном).
ПЛАЗМА представляет собой газ, в котором находятся атомы в ионизированном состоянии.
Плазма образуется в результате индукционного нагрева газа (аргона), протекающего через систему кварцевых трубок. Трубки размещены внутри рабочей катушки ВЧ - генератора, представляющей собой медную спираль (индуктор), обвивающую верхнюю часть трубок.
302
Приложение 29
Формы электродов для АЭСА
Приложение 30
Разложение излучения в спектр
Разложение света призмой основано на зависимости угла отклонения луча, прошедшего через призму, от показателя преломления, различного для разных длин волн.
Дифракционная решетка разлагает падающий на решетку пучок света по длинам волн, то есть в спектр.
303

Приложение 31
Лист атласа дугового спектра железа
304
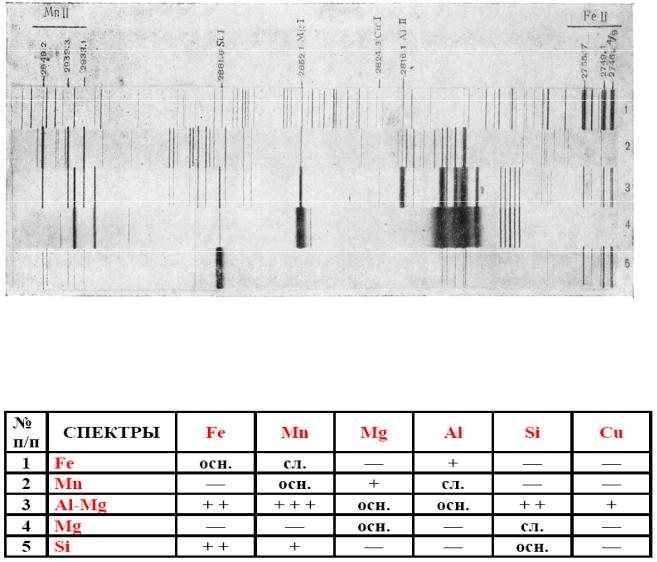
Приложение 32
Качественный спектральный анализ с помощью сопоставления спектров анализируемых объектов со спектром железа
1 – спектр железа, 2 – спектр марганца, 3 – спектр алюминиево-магниевого сплава, 4 – спектр чистого магния, 5 – спектр кремния
305

Приложение 33
Фрагмент записи результатов квантометрического анализа оценки качества алюминия
306

Приложение 34
Спектрометры с индуктивно связанной плазмой
307

Приложение 35
Лазерный атомно-эмиссионный спектрометр
308
|
|
|
|
|
Приложение 36 |
|
Метрологические характеристики метода атомно-эмиссионной |
||||||
|
спектроскопии |
|
|
|
|
|
|
|
|
|
|
|
|
Способ |
Воспр., |
|
ПрО, %, масс. |
|
t, мин |
|
регистрации |
Sr , % |
|
|
|
|
|
ФОТОГРАФИЧЕСКИЙ |
5 |
|
|
|
|
25-30 минут |
|
|
|
|
|
|
на 10-12 |
Дуга |
|
|
10 6 |
10 4 |
|
|
|
|
|
|
|
|
элементов |
Искра |
|
|
10 3 |
10 1 |
|
|
ФОТОЭЛЕКТРИЧЕСКИЙ |
|
|
|
|
|
1-2 минуты |
|
|
|
|
|
|
на 10-30 |
Дуга |
0,5 |
|
10 4 |
10 2 |
|
|
|
|
|
|
|
|
элементов |
Искра |
0,2 |
|
|
|
|
|
ИСП |
0,01 – 0,05 |
|
10 8 |
10 2 |
|
|
Пламя |
2 – 4 |
|
10 4 |
10 2 |
|
|
Приложение 37
Хроматография
Анализ сложной смеси веществ часто требует разделения ее на составляющие. Такие широкоизвестные методы разделения, как дистилляция, кристаллизация, экстракция и адсорбция основаны на фазовых равновесиях. В этих процессах молекулы веществ, образующих смесь, переходят через границу раздела, стремясь к такому распределению между фазами, при котором в каждой из них устанавливается равновесная концентрация.
Если свойства составляющих исследуемой смеси близки, то достаточная степень разделения достигается лишь многократным повторением элементарного акта разделения. Если разделение смеси проводить в таких условиях, когда одна из фаз (подвижная) перемещается относительно другой (неподвижной), то разделение молекул осуществляется благодаря постоянному перемещению подвижной фазы. Как и при фазовом равновесии, молекулы, выходящие из подвижной фазы, возвращаются в нее после взаимодействия с неподвижной фазой, попадая в новый элемент объема подвижной фазы. Таким образом, процесс разделения повторяется многократно, что позволяет получить высокую эффективность разделения.
309
Итак, хроматографический метод – это физико-химический метод
разделения и определения компонентов сложных смесей газов, паров, жидкостей или растворенных веществ, основанный на использовании сорбционных процессов в динамических условиях.
Воснове хроматографического разделения смесей веществ лежит способность их компонентов к различному распределению между двумя несмешивающимися фазами - подвижной и неподвижной.
Впростейшем варианте эти условия осуществляются при прохождении раствора, содержащего растворенные вещества, через колонку со слоем сорбента. В основе хроматографического разделения лежит различие в сорбционной активности компонентов смеси по отношению к данному сорбенту.
Сорбция (sorbeo - поглощаю (лат.)) – это процесс поглощения твердым телом или жидкостью (сорбентом) газообразного или растворенного вещества (сорбата). Обратный процесс называется десорбцией.
Виды сорбции:
1)адсорбция – поглощение вещества (абсорбата) поверхностью твердого или жидкого адсорбента, т.е. концентрирование вещества из объема фаз на по-
верхности раздела между ними;
2)абсорбция - поглощение веществ из газа или жидкости жидкостями (реже твердыми телами), которое происходит во всем объеме поглотителя;
3)хемосорбция - поглощение веществ жидким или твердым сорбентом с образованием химического соединения;
4)капиллярная конденсация – образование жидкой фазы в порах и капиллярах твердого сорбента при поглощении паров веществ. Хроматографические методы классифицируются различными способами
(табл. 5) в зависимости от агрегатного состояния фаз, типа взаимодействия (механизма или химизма процесса), аппаратуры или техники проведения хроматографического процесса.
310
Таблица 5
ОСНОВНЫЕ ВИДЫ ХРОМАТОГРАФИИ
Вид хромато- |
Подвиж- |
Неподвиж- |
Форма |
Механизм |
графии |
ная фаза |
ная фаза |
разделения |
|распределения |
Газовая: |
Газ |
Твердая |
Колонка |
Адсорбцион- |
Газо- |
||||
адсорбционная |
|
|
|
ный |
Газожидкост- |
Газ |
Жидкость |
Колонка |
Распредели- |
ная |
|
|
|
тельный |
Жидкостная: |
Жидкость |
Твердая |
Колонка |
Адсорбцион- |
твердожидко- |
||||
стная |
|
|
|
ный |
жидкостно- |
Жидкость |
Жидкость |
Колонка |
Распределитель- |
жидкостная |
|
|
|
ный |
ионообменная |
Жидкость |
Твердая |
Колонка |
Ионный обмен |
Тонкослойная |
Жидкость |
Твердая |
Тонкий |
Адсорбцион- |
|
|
|
слой |
ный |
|
Жидкость |
Жидкость |
Тонкий |
Распределитель- |
|
|
|
Слой |
ный |
|
|
|
|
|
Бумажная |
Жидкость |
Жидкость |
Лист |
Распределитель- |
|
|
|
бумаги |
ный |
|
|
|
|
|
Ситовая (гель- |
Жидкость |
Жидкость |
Колонка |
По размерам |
проникающая) |
|
|
|
молекул |
|
|
|
|
|
Для разделения разных молекул неподвижная фаза должна обладать хотя бы одним из следующих свойств: способностью к физическому сорбированию вещества, находящегося в подвижной фазе; способностью к химическому сорбированию вещества подвижной фазы; способностью к растворению разделяемых веществ; наличием пористой структуры, способной удерживать одни вещества и не задерживать другие в зависимости от их размеров или формы.
По механизму взаимодействия сорбента и сорбата выделяют несколько видов хроматографии.
Если неподвижная фаза представляет собой твердое вещество, способное к
311
адсорбции определяемого вещества, то такая хроматографическая система на-
зывается адсорбционной.
В распределительной хроматографии неподвижной фазой является жидкость, в которой способно растворяться анализируемое вещество. Разделение основано на различии в растворимости разделяемых веществ в неподвижной фазе или на различии в растворимости веществ в подвижной и неподвижной жидкой фазах.
Элюентная хроматография
Элюент - это подвижная фаза, которую вводят в слой неподвижной фазы. Элюат - это подвижная фаза, которая выходит из колонки и содержит разделенные компоненты. В элюате тем или иным способом определяют содержа-
ние компонентов.
Элюентная хроматография является наиболее распространенной в современной хроматографии. Сначала хроматографическую колонку промывают элюентом (раствором или растворителем), который должен обладать меньшей сорбируемостью, чем все разделяемые вещества. Затем в поток элюента вводят разделяемые вещества и продолжают непрерывное пропускание элюента через колонку. Разделяемые вещества в соответствии с их сорбируемостью перемещаются вдоль колонки с разными скоростями и в результате выходят из колонки поочередно, разделенные зонами чистого элюента. При этом первым появляется наименее сорбируемый компонент. Хроматограмма представляет собой несколько пиков, имеющих форму гауссовой кривой.
Аппаратура и техника выполнения хроматографического анализа
Блоксхема газового хроматографа
Блок-схема хроматографа состоит из следующих основных узлов:
1.Система подачи подвижной фазы (газовый баллон, насос для подачи жидкой подвижной фазы).
2.Дозатор.
3.Колонка.
4.Детектор.
5.Регистратор (самописец, интегратор, ЭВМ).
6.Компьютер для обработки информации.
312
Основные типы детекторов для решения поставленных нами задач являются катарометр, пламенно-ионизационный детектор, масс-спектрометр.
Подробно смотри: типы детекторов [2], кн. 1, с. 303…306.
Хроматографические параметры
Хроматограмма - это графическое изображение процесса распределения веществ в элюате.
Основными параметрами хроматограмм являются:
- время удерживания tR - которое определяют по стандартным веществам, оно является основой идентификации (качественного определения) веществ на хроматограмме;
- площадь хроматографического пика позволяет провести количественное определение компонента, поскольку этот параметр пропорционален концентрации или количеству вещества в хроматографической зоне.
Подробно рассмотрены в [2], кн. 1, с. 292…294.
313
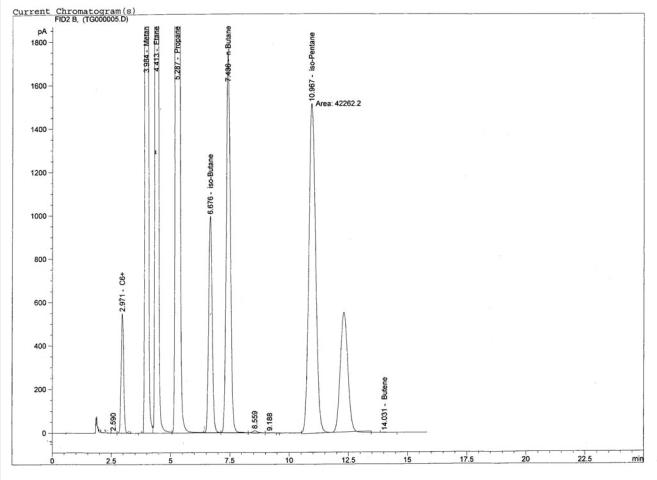
Приложение 38
Фрагмент хроматограммы
314
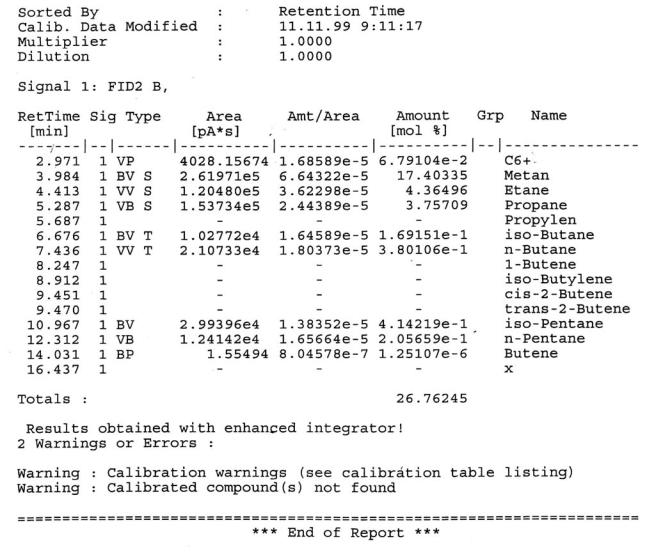
Приложение 39
Компьютерная расшифровка хроматограммы
315

Приложение 40
Лабораторный газовый хроматограф Хромос
Особенности:
–Использование насадочных и капиллярных колонок;
–Стандартный набор детекторов: ПИД, ТИД, ЭЗД, ДТП;
–Электронная система регулирования газовых потоков;
–Возможность использования одного компьютера для нескольких хроматографов.
316
Приложение 41
Автоматизация анализа. Компьютеризация анализа. Организация аналитического контроля
При изучении этого раздела рассмотрите следующие вопросы:
1.Лабораторные роботы, автоматический анализ.
2.Использование компьютеров для управления аналитическими приборами: управление и сбор данных; первичная обработка данных.
3.Внедрение лабораторно-информационных систем.
Автоматизация анализа - одно из основных направлений развития аналитической химии и аналитического контроля производства. К автоматизации очень тесно примыкает компьютеризация как отдельных аналитических приборов, так и всего аналитического цикла. Автоматизация возможна на разных стадиях анализа. Проще всего на стадии измерения аналитического сигнала и обработки результатов. Наиболее сложными и не всегда разрешимыми на данном этапе являются вопросы, связанные с отбором пробы и подготовкой ее к анализу.
В последнее десятилетие началось активное внедрение лабораторно-
информационных систем (ЛИС, LIMS-Laboratory Information Management Systems) в практику работы аналитической сервисной службы предприятий.
Деятельность аналитических подразделений на любом предприятии связана с проведением большого количества аналитических испытаний различных объектов, составлением разнообразных аналитических отчетов в соответствии с требованиями различных нормативных документов (ГОСТ, ОСТ, ТУ, СТП). На любом предприятии аналитический контроль включает:
входной контроль сырья,
контроль промежуточных продуктов по ходу технологического процесса,
контроль качества полученной продукции.
Это огромный объем ежедневной информации.
Чтобы обеспечить получение достоверной информации, необходимо учитывать ряд моментов:
317
1.Строго контролируемым должно быть соблюдение всех требований к качеству проведения анализов, включая качество измерения аналитического сигнала и проведения обработки полученной информации, оговоренных в соответствующих нормативных документах (ГОСТ, ОСТ, ТУ, СТП).
2.В любой отрасли промышленности и в системе организации охраны окружающей среды существуют определенные нормы качественных характеристик анализируемого объекта ( в промышленности это качественные характеристики состава сырья, полупродуктов и конечных продуктов; в системе организации охраны окружающей среды – это предельно допустимые концентрации); эти нормы также закреплены в соответствующей документации и результаты испытаний должны соответствовать этим нормам.
3.Четкое функционирование аналитической службы основывается на использовании широкого ассортимента лабораторного оборудования, средств измерений и приборов, которые должны проходить периодические метрологические оценки, профилактическое обслуживание и проверку калибровочных процедур.
4.Достоверность полученной информации напрямую зависит от квалификации исполнителей.
Лаборатория, выполняющая сертификационные анализы, должна следовать системе контроля качества согласно ISO 17025 (2005 г.), поддерживая соответствующий уровень компетенции лаборатории. Система достаточно сложная, требующая подготовки очень большого количества регламентирующих документов (методик, обязанностей персонала, принципов поддержания системы качества) и разного рода форм с определенной системой кодификации.
Для автоматизации заполнения форм, хранения и обработки результатов анализов, систематизированного хранения библиотек документов (их версий) на современном рынке аналитического оборудования представлены системы обработки лабораторной информации (Laboratory Information Management Systems – LIMS/ ЛИМС).
Развитие ЛИМС началось примерно 25 лет назад с предложения отдельных компонентов системы. Сейчас это развитая сетевая клиент-серверная архитектура, обеспечивающая интегрирование ЛИМС с системой управления пред-
приятием (ERP: Enterprise Resourse Planning). Центральным элементом ЛИМС
318
является реляционная база данных ( обычно SQL – сервер), обрамленная сервисами, обеспечивающими функционирование системы на основе рекомендаций ГОСТ Р – ISO/IEC 17025:2005 и некоторых других. С основными принципами построения и функционирования ЛИМС можно ознакомиться на примере сис-
темы LabWare www.labware.ru .
Таким образом, предлагаемые ЛИМС имеют открытую архитектуру и могут встраиваться в более общую систему менеджмента предприятия ( управление, логистика и т. д. ). Предприятие, декларирующее производство согласно требованиям ISO-9000 (что является основой для успешного продвижения продукции на рынке), автоматически должно следовать и требованиям ISO – 17025.
Подробно материал рассматривается в [2], кн. 2. с. 404…446.
319
|
СОДЕРЖАНИЕ |
|
1. |
ИНФОРМАЦИЯ О ДИСЦИПЛИНЕ..................................................................................................... |
4 |
|
1.1 . ПРЕДИСЛОВИЕ............................................................................................................................... |
4 |
|
1.2 . СОДЕРЖАНИЕ ДИСЦИПЛИНЫ И ВИДЫ УЧЕБНОЙ РАБОТЫ............................................. |
6 |
|
1.2.1. СОДЕРЖАНИЕ ДИСЦИПЛИНЫ ПО ГОС ............................................................................... |
6 |
|
1.2.2. ОБЪЕМ ДИСЦИПЛИНЫ И ВИДЫ УЧЕБНОЙ РАБОТЫ....................................................... |
7 |
|
1.2.3. ПЕРЕЧЕНЬ ВИДОВ ПРАКТИЧЕСКИХ ЗАНЯТИЙ И КОНТРОЛЯ....................................... |
7 |
2. |
РАБОЧИЕ УЧЕБНЫЕ МАТЕРИАЛЫ.................................................................................................. |
8 |
|
2.1. РАБОЧАЯ ПРОГРАММА .................................................................................................................. |
8 |
|
2.2. ТЕМАТИЧЕСКИЙ ПЛАН ДИСЦИПЛИНЫ................................................................................... |
17 |
|
2.3. СТРУКТУРНО-ЛОГИЧЕСКАЯ СХЕМА ДИСЦИПЛИНЫ .......................................................... |
23 |
|
2.4. ВРЕМЕННОЙ ГРАФИК ИЗУЧЕНИЯ ДИСЦИПЛИНЫ ПРИ ИСПОЛЬЗОВАНИИ....................... |
|
|
ИНФОРМАЦИОННО-КОММУНИКАЦИОННЫХ ТЕХНОЛОГИЙ.................................................. |
24 |
|
2.5. ПРАКТИЧЕСКИЙ БЛОК.................................................................................................................. |
25 |
|
2.5.1. ЛАБОРАТОРНЫЙ ПРАКТИКУМ............................................................................................ |
25 |
|
2.6. БАЛЛЬНО-РЕЙТИНГОВАЯ СИСТЕМА ОЦЕНКИ ЗНАНИЙ ................................................... |
28 |
3. ИНФОРМАЦИОННЫЕ РЕСУРСЫ ДИСЦИПЛИНЫ........................................................................... |
29 |
|
|
3.1. БИБЛИОГРАФИЧЕСКИЙ СПИСОК............................................................................................... |
29 |
|
3.2. ОПОРНЫЙ КОНСПЕКТ................................................................................................................... |
30 |
|
3.3. ГЛОССАРИЙ.................................................................................................................................... |
79 |
|
3.4. ТЕХНИЧЕСКИЕ И ПРОГРАММНЫЕ СРЕДСТВА ОБЕСПЕЧЕНИЯ ДИСЦИПЛИНЫ........... |
92 |
|
3.5. МЕТОДИЧЕСКИЕ УКАЗАНИЯ К ВЫПОЛНЕНИЮ ЛАБОРАТОРНЫХ РАБОТ................... |
93 |
|
ЛАБОРАТОРНАЯ РАБОТА №1........................................................................................................ |
96 |
|
ЛАБОРАТОРНАЯ РАБОТА №2...................................................................................................... |
102 |
|
ЛАБОРАТОРНАЯ РАБОТА №3...................................................................................................... |
111 |
|
ЛАБОРАТОРНАЯ РАБОТА №4...................................................................................................... |
114 |
|
ЛАБОРАТОРНАЯ РАБОТА №5...................................................................................................... |
120 |
|
ЛАБОРАТОРНАЯ РАБОТА №6...................................................................................................... |
130 |
|
ЛАБОРАТОРНАЯ РАБОТА №7...................................................................................................... |
135 |
|
ЛАБОРАТОРНАЯ РАБОТА №8...................................................................................................... |
141 |
|
ЛАБОРАТОРНАЯ РАБОТА №9...................................................................................................... |
155 |
|
ЛАБОРАТОРНАЯ РАБОТА №10.................................................................................................... |
159 |
|
ЛАБОРАТОРНАЯ РАБОТА №11.................................................................................................... |
168 |
4. БЛОК КОНТРОЛЯ ОСВОЕНИЯ ДИСЦИПЛИНЫ ............................................................................. |
170 |
|
|
4.1. ОБЩИЕ УКАЗАНИЯ....................................................................................................................... |
170 |
|
4.2. ЗАДАНИЕ НА КОНТРОЛЬНУЮ РАБОТУ И МЕТОДИЧЕСКИЕ УКАЗАНИЯ К ЕЕ |
|
|
ВЫПОЛНЕНИЮ..................................................................................................................................... |
170 |
|
4.2.1. ОБЩИЕ УКАЗАНИЯ.............................................................................................................. |
170 |
|
4.2.2. ЗАДАНИЕ НА КОНТРОЛЬНУЮ РАБОТУ......................................................................... |
172 |
|
4.2.3. МЕТОДИЧЕСКИЕ УКАЗАНИЯ К ВЫПОЛНЕНИЮ КОНТРОЛЬНОЙ РАБОТЫ ......... |
187 |
|
4.3. ТЕКУЩИЙ КОНТРОЛЬ. ТРЕНИРОВОЧНЫЕ ТЕСТЫ.............................................................. |
252 |
|
4.4 ИТОГОВЫЙ КОНТРОЛЬ ................................................................................................................ |
258 |
ПРАВИЛЬНЫЕ ОТВЕТЫ НА ТРЕНИРОВОЧНЫЕ ТЕСТЫ................................................................. |
259 |
|
ПРИЛОЖЕНИЯ........................................................................................................................................... |
260 |
|
320
Беляева Татьяна Валентиновна
Химия, ч. 3. Аналитическая химия
Учебно-методический комплекс
Редактор Т. В. Шабанова Сводный темплан 2009 г.
ЛР № 020308 от 14.02.97
Подписано в печать 00.02.2010. Формат 60х84 1/16 Б. кн.-журн. П.л. 20,00 . Б.л. 10,00. Издательство СЗТУ.
Тираж 150. Заказ Северо-Западный государственный заочный технический университет
Издательство СЗТУ, член Издательско-полиграфической ассоциации университетов России 191186, Санкт-Петербург, ул. Миллионная, д.5
321