

Biomedical EPR Part-B Methodology Instrumentation and Dynamics - Sandra R. Eaton
.pdf90 DEVKUMAR MUSTAFI AND MARVIN W. MAKINEN
with macromolecules ranges from ordered and essentially static, detectable by X-ray and neutron diffraction methods at ambient temperature over periods of days, to that of bulk solvent with rapid interconversion 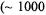
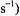 of structural arrangements and reorientation of hydrogen-bonds. Special interest has long been directed towards sites in macromolecules with structured water stabilized through hydrogen-bonding. Such sites may underlie the catalytic function of enzymes whereby water molecules are strategically positioned to react as nucleophiles or to facilitate rapid translocation of protons in clusters through hydrogen-bonding and electrostatic interactions.
of structural arrangements and reorientation of hydrogen-bonds. Special interest has long been directed towards sites in macromolecules with structured water stabilized through hydrogen-bonding. Such sites may underlie the catalytic function of enzymes whereby water molecules are strategically positioned to react as nucleophiles or to facilitate rapid translocation of protons in clusters through hydrogen-bonding and electrostatic interactions.
To assign the chemical role of water molecules strategically positioned in such sites requires measurement of their atomic coordinates and description of their structural interactions with protein residues. This objective, for enzyme reaction intermediates, requires preparation of the enzyme in a catalytically functional state and application of a physical method to determine atomic positions. Because the lifetimes of catalytically competent enzyme reaction intermediates are much shorter than the time of data collection, X-ray methods have generally taken advantage of the stability of enzyme-inhibitor complexes to mimic enzyme-substrate interactions. However, non-productive structural relationships are responsible for the long-term stability of enzyme-inhibitor complexes in contrast to the labile, short-lived, productive enzyme-substrate interactions responsible for the catalytic conversion of substrate to product. For this reason, enzymeinhibitor complexes can offer only a partial description of the catalytically competent structure of the active site. Furthermore, introduction of inhibitor ligands into enzyme active sites is often associated with displacement of strategically positioned water molecules. This chapter describes how these drawbacks can be avoided by combined application of electron nuclear double resonance (ENDOR) spectroscopy with cryoenzymology to isolate catalytically competent enzyme reaction intermediates for structural analysis.
The underlying physical process of interest in the application of ENDOR spectroscopy is hyperfine (hf) coupling, the interaction between an unpaired electron with a nearby magnetic nucleus. ENDOR provides the most precise method to measure the influence of the (oscillating) magnetic field produced by the unpaired electron on the nucleus, resulting in a shift in its resonance frequency from that in the absence of the electron. This measurement can lead to highly precise estimates of the separation between the unpaired electron and the nucleus and provides thereby a sensitive method to determine electronic and molecular structure. In this review we describe ENDOR studies from our laboratory that have led to assignments of the coordination geometry of metal-bound or hydrogen-bonded solvent in small
ANGLE-SELECTED ENDOR |
91 |
molecules, nucleotide complexes, and proteins. We emphasize instances in which comparison of the details of small molecule systems with their counterparts in proteins and enzymes has brought improved understanding of macromolecular function. We also discuss briefly how extensions of this spectroscopic approach may enhance structural analysis and thereby bring new insights into the study of structure-function relationships of biological macromolecules.
For further discussions of ENDOR and related double resonance methods, the reader should consult Atherton (1993), whose textbook on electron paramagnetic resonance (EPR) places a heavy emphasis on ENDOR in comparison to others, or specialized monographs on other forms of double resonance spectroscopy (Kevan and Kispert, 1976; Dorio and Freed, 1979; Kurreck et al., 1988). Ch. 15 of volume 23 describes electron spin-echo envelope modulation (ESEEM) spectroscopy, a related electron magnetic resonance method complementary to ENDOR. Since ENDOR is usually applied to determine dipolar electron-nucleus distances between paramagnetic sites and nearby magnetic nuclei, Ch. 8, describing applications of pulsed EPR for electron-electron distance measurements, should be also consulted. Both continuous wave (cw) and pulsed ENDOR spectrometers have been commercially available for several decades. Studies described from our laboratory have been carried out with a cw X-band Bruker ESP 300E spectrometer equipped with a  cylindrical cavity, Bruker ENDOR accessory, and Oxford Instruments ESR 910 liquid helium cryostat.
cylindrical cavity, Bruker ENDOR accessory, and Oxford Instruments ESR 910 liquid helium cryostat.
Since the first demonstration of ENDOR to measure hf coupling of 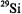 nuclei with impurity sites in silicon (Feher, 1956, 1957), it has been widely applied to free radicals in solution and in the solid state. Moreover, its application has resulted in precise determination of the coordinates of protons and deuterons of metal-bound and hydrogen-bonded water molecules in single crystals of diamagnetic metal ion complexes into which
nuclei with impurity sites in silicon (Feher, 1956, 1957), it has been widely applied to free radicals in solution and in the solid state. Moreover, its application has resulted in precise determination of the coordinates of protons and deuterons of metal-bound and hydrogen-bonded water molecules in single crystals of diamagnetic metal ion complexes into which
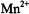 (van Ormondt and Visser, 1968),
(van Ormondt and Visser, 1968),  (Hutchison and McKay, 1977; Fields and Hutchison, 1985),
(Hutchison and McKay, 1977; Fields and Hutchison, 1985), 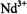 (Hutchison and Orlowski, 1980), and
(Hutchison and Orlowski, 1980), and 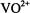 (Atherton and Shackleton, 1980, 1984) have been incorporated as the paramagnetic probe. In these studies the uncertainty in determining coordinates of protons and deuterons was 0.002-0.099 Å for metal-nucleus distances over a 2-5 Å range. The major source of error was the uncertainty in determining the direction of the laboratory magnetic field in the crystal. This precision compares favorably with that achieved in high resolution X- ray diffraction studies of small molecules in which the positions of hydrogen atoms are directly determined on the basis of scattering data after extensive crystallographic least-squares refinement. ENDOR of single crystals containing substitutionally dilute paramagnetic species has been also applied to determine positions of
(Atherton and Shackleton, 1980, 1984) have been incorporated as the paramagnetic probe. In these studies the uncertainty in determining coordinates of protons and deuterons was 0.002-0.099 Å for metal-nucleus distances over a 2-5 Å range. The major source of error was the uncertainty in determining the direction of the laboratory magnetic field in the crystal. This precision compares favorably with that achieved in high resolution X- ray diffraction studies of small molecules in which the positions of hydrogen atoms are directly determined on the basis of scattering data after extensive crystallographic least-squares refinement. ENDOR of single crystals containing substitutionally dilute paramagnetic species has been also applied to determine positions of 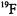 (Baker et al., 1968),
(Baker et al., 1968), 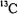 (Rudin et al., 1982),
(Rudin et al., 1982),

92 |
|
DEVKUMAR MUSTAFI AND MARVIN W. MAKINEN |
and |
and |
(Feher et al., 1973; Scholes et al., 1982) with comparable |
precision.
While the precision achieved through single crystal ENDOR analysis is impressive, its application to biological problems is limited. The usual dimensions of crystals of biological macromolecules and concentration of paramagnetic sites place severe limitations on signal-to-noise and the total number of spins in the spectrometer cavity. Also, the systems of interest, e.g., macromolecular complexes or intermediates of enzyme-catalyzed reactions, may not be obtainable in single crystal form. For these reasons, a more conventional approach has been applied with use of polycrystalline or frozen glassy samples in which EPR spectra reflect a “powder” average over all molecular orientations with respect to the laboratory magnetic field.
The orientation dependence of magnetic hyperfine interactions was first described for EPR spectra of crystals and powder samples of free radicals in the absence of g anisotropy (Blinder, 1960). Subsequently Rist and Hyde (1968, 1970) demonstrated in the case of g anisotropy that, at some settings of the magnetic field, the so-called “turning points” in the EPR spectrum, “single-crystal-type” ENDOR spectra can be obtained from molecules essentially in a single orientation. Hoffman and coworkers (1984) presented a general formulation of the orientation dependence of ENDOR spectra of polycrystalline samples for which g anisotropy dominates. Furthermore, it has been shown that ENDOR spectra can be obtained throughout the EPR spectrum when frequency modulation of the swept radiofrequency (rf) field is employed (Hurst et al., 1985). Thus, one is not confined only to the limited number of specific orientations found at the turning points of the EPR absorption spectrum.
The first applications of “angle-selected ENDOR” to determine structure and to assign relative coordinates of protons with respect to the paramagnetic center were published for polycrystalline dithiophosphate and dimethyldithiocarbamate complexes of nickel(II) doped with  (Yordanov and Zdravkova, 1986; Yordanov et al., 1986), and for
(Yordanov and Zdravkova, 1986; Yordanov et al., 1986), and for  adducts of Schiff bases (Attanasio, 1986, 1989) and
adducts of Schiff bases (Attanasio, 1986, 1989) and  (Yim
(Yim
and Makinen, 1986) in |
frozen glassy solutions. At about the same time, |
insightful descriptions |
of the mathematical theory underlying the |
interpretation of ENDOR spectra of randomly oriented molecules appeared from the laboratories of Kreilick (Henderson et al., 1985; Hurst et al., 1985) and Hoffman (Hoffman et al., 1984,1985; Hoffman and Gurbiel, 1989). The methodology has been subsequently extended in our laboratory to a variety of complexes (Mustafi and Makinen, 1988; Mustafi et al., 1992; Mustafi and Nakagawa, 1994, 1996; Makinen and Mustafi, 1995; Jiang and Makinen, 1995; Mustafi et al., 2000; Makinen and Brady, 2002) and nitroxyl spin-labels (Wells and Makinen, 1988; Mustafi et al., 1990b,
complexes (Mustafi and Makinen, 1988; Mustafi et al., 1992; Mustafi and Nakagawa, 1994, 1996; Makinen and Mustafi, 1995; Jiang and Makinen, 1995; Mustafi et al., 2000; Makinen and Brady, 2002) and nitroxyl spin-labels (Wells and Makinen, 1988; Mustafi et al., 1990b,
ANGLE-SELECTED ENDOR |
93 |
1990c, 1993, 1995, 1997, 2001, 2002; Wells et al., 1990; Mustafi and Makinen, 1995; Jiang et al., 1998; Makinen, 1998; Makinen et al., 1998) to determine small molecule and macromolecular structure. The uncertainty in electron-nucleus distances estimated from ENDOR line widths in these studies is  over an approximate 3-8 Å range for
over an approximate 3-8 Å range for 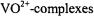 (Makinen and Mustafi, 1995) and 4-11
(Makinen and Mustafi, 1995) and 4-11  range for spin-labels (Makinen et al., 1998).
range for spin-labels (Makinen et al., 1998).
2.ENDOR ASSIGNMENT OF MOLECULAR
STRUCTURE AND CONFORMATION WITH 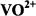 AND NITROXYL SPIN-LABELS
AND NITROXYL SPIN-LABELS
2.1EPR Absorption Dependent upon Molecular Orientation
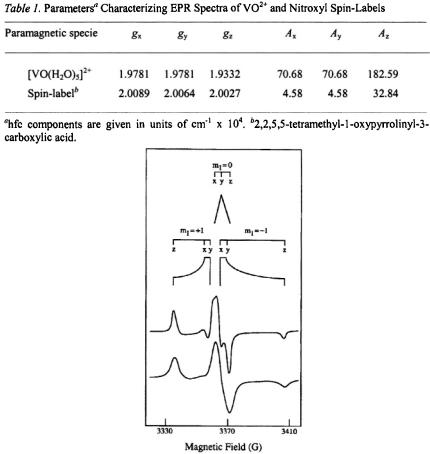
The physical process underlying ENDOR spectroscopy is the hf interaction between an unpaired electron and a magnetic nucleus. This is due to the interaction of the unpaired electron spin of the paramagnetic site with nearby magnetic nuclei, producing shifts in their resonance frequencies. This phenomenon gives rise to broadening of EPR spectra, as illustrated in Figs. 1 and 2, by comparison of the spectra of 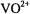 in protiated and perdeuterated methanol and of the nitroxyl spin-label 2,2,5, 5-tetramethyl-1-oxypyrroline- 3-carboxylic acid in its protiated and perdeuterated forms, respectively. In Fig. 1 the EPR absorption features of
in protiated and perdeuterated methanol and of the nitroxyl spin-label 2,2,5, 5-tetramethyl-1-oxypyrroline- 3-carboxylic acid in its protiated and perdeuterated forms, respectively. In Fig. 1 the EPR absorption features of 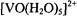 are broader in a solvent of natural abundance isotope composition than in the perdeuterated solvent, reflecting the stronger but unresolved electron-nuclear hf couplings of nearby protons of solvent molecules in the inner and outer coordination shells. On the other hand, the line broadening of nitroxyl spin-labels, as seen in Fig. 2, is entirely dominated by the covalent hydrogens in the pyrrolinyl ring and the methyl groups adjacent to the N–O group. There is no perceptible change in EPR line width of the spin-label with change of solvent. These examples illustrate the major limitation of EPR in probing structural details of a paramagnetic site, namely, the failure to resolve ligand hf or superhyperfine (shf) couplings of nearby magnetic nuclei that interact with the unpaired electron spin. Nonetheless, quantitative evaluation of shf couplings is essential for structure determination. In order to assign nuclear coordinates and to estimate electron-nucleus separations, it is necessary to measure the strength of shf interactions and to determine their relationships to magnetic axes in the molecule and to the external, static laboratory magnetic field
are broader in a solvent of natural abundance isotope composition than in the perdeuterated solvent, reflecting the stronger but unresolved electron-nuclear hf couplings of nearby protons of solvent molecules in the inner and outer coordination shells. On the other hand, the line broadening of nitroxyl spin-labels, as seen in Fig. 2, is entirely dominated by the covalent hydrogens in the pyrrolinyl ring and the methyl groups adjacent to the N–O group. There is no perceptible change in EPR line width of the spin-label with change of solvent. These examples illustrate the major limitation of EPR in probing structural details of a paramagnetic site, namely, the failure to resolve ligand hf or superhyperfine (shf) couplings of nearby magnetic nuclei that interact with the unpaired electron spin. Nonetheless, quantitative evaluation of shf couplings is essential for structure determination. In order to assign nuclear coordinates and to estimate electron-nucleus separations, it is necessary to measure the strength of shf interactions and to determine their relationships to magnetic axes in the molecule and to the external, static laboratory magnetic field 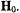 This objective is best achieved by application of ENDOR spectroscopy.
This objective is best achieved by application of ENDOR spectroscopy.

94 |
DEVKUMAR MUSTAFI AND MARVIN W. MAKINEN |
Figure 1. First-derivative EPR absorption spectra of 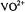 in 50:50 (v/v) (a)
in 50:50 (v/v) (a)  and (b)
and (b) 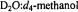 solvent mixtures. The parallel
solvent mixtures. The parallel 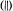 and perpendicular
and perpendicular 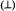 EPR absorption components (–7/2, –5/2, · · · , +5/2, +7/2) are identified in sequence in the lowto high-field direction. The double-headed arrow indicates the magnetic field position corresponding to the forbidden
EPR absorption components (–7/2, –5/2, · · · , +5/2, +7/2) are identified in sequence in the lowto high-field direction. The double-headed arrow indicates the magnetic field position corresponding to the forbidden  transition. The spectra were obtained with a Bruker ER200D spectrometer operating at 9.5 GHz with the sample temperature maintained at 10 K with an Oxford Instruments 910 ESR liquid helium cryostat. Reprinted from Mustafi and Makinen (1988) with permission by the American Chemical Society.
transition. The spectra were obtained with a Bruker ER200D spectrometer operating at 9.5 GHz with the sample temperature maintained at 10 K with an Oxford Instruments 910 ESR liquid helium cryostat. Reprinted from Mustafi and Makinen (1988) with permission by the American Chemical Society.
In Table 1 we compare the spectroscopic parameters describing the principal components of the 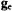 tensor and nuclear hyperfine tensor of the
tensor and nuclear hyperfine tensor of the 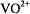 ion and of the N–O group of nitroxyl spin-labels. For both paramagnetic species, each with one unpaired electron, EPR and ENDOR spectra can be described by the spin Hamiltonian in Eq. (1), which includes the electronic Zeeman interaction
ion and of the N–O group of nitroxyl spin-labels. For both paramagnetic species, each with one unpaired electron, EPR and ENDOR spectra can be described by the spin Hamiltonian in Eq. (1), which includes the electronic Zeeman interaction 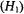 and the nuclear Zeeman interaction
and the nuclear Zeeman interaction 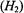 while hf interactions of the unpaired electron with different classes of nuclei are contained within
while hf interactions of the unpaired electron with different classes of nuclei are contained within 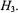 In Eq. (1),
In Eq. (1),  and
and  represent the Bohr electron and nuclear magneton,
represent the Bohr electron and nuclear magneton,  the electronic Zeeman interaction tensor,
the electronic Zeeman interaction tensor,  the nuclear g-factor,
the nuclear g-factor,  the external (laboratory) magnetic field, S and I the electron and nuclear spin operators, respectively, and A the electronnucleus hf tensor.
the external (laboratory) magnetic field, S and I the electron and nuclear spin operators, respectively, and A the electronnucleus hf tensor.
ANGLE-SELECTED ENDOR |
95 |
Figure 2. Schematic diagram of the rigid limit EPR powder pattern for the first-derivative EPR absorption spectra of methyl N-(2,2,5,5-tetramethyl-1-oxypyrrolinyl-3-carboxyl)-L-ala- ninate and of methyl 
 in frozen
in frozen  In the upper half of the figure, stick diagrams identify different components of the electronic spin transitions according to
In the upper half of the figure, stick diagrams identify different components of the electronic spin transitions according to  and
and 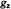 and values of
and values of 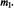 The upper spectrum is of the
The upper spectrum is of the 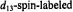 compound while the lower spectrum is of the spinlabeled compound of natural abundance isotope composition. EPR parameters used to calculate the stick diagrams at a microwave frequency of 9.45 GHz are given in Table 1. Reprinted from Makinen et al. (1998) with permission.
compound while the lower spectrum is of the spinlabeled compound of natural abundance isotope composition. EPR parameters used to calculate the stick diagrams at a microwave frequency of 9.45 GHz are given in Table 1. Reprinted from Makinen et al. (1998) with permission.
As a rapidly tumbling species in fluid solution at ambient temperature, the  ion gives rise to eight sharp EPR transitions because of the hf interaction of the unpaired electron with the 100% naturally abundant (I = 7/2)
ion gives rise to eight sharp EPR transitions because of the hf interaction of the unpaired electron with the 100% naturally abundant (I = 7/2) 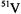 nucleus. Correspondingly, the nitroxyl radical gives rise to three EPR transitions due to the interaction of the unpaired spin with the (I = 1)
nucleus. Correspondingly, the nitroxyl radical gives rise to three EPR transitions due to the interaction of the unpaired spin with the (I = 1) 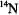 nucleus. In the limit of a poly-crystalline sample or frozen glassy
nucleus. In the limit of a poly-crystalline sample or frozen glassy
96 DEVKUMAR MUSTAFI AND MARVIN W. MAKINEN
solution near the temperature of liquid helium, as illustrated in Figs. 1 and 2, the EPR spectra are accordingly composites from different sets of molecules
designated according to the projections of the |
or |
nuclear moments |
|
onto the laboratory magnetic field |
For both unpaired spin systems, the |
||
separation of the principal components of the |
tensor as a result of |
||
anisotropic hf interactions provides the basis to apply angle-selected
ENDOR (Rist and Hyde, 1970). |
|
|
||
For |
with an axially symmetric |
tensor and an axially symmetric |
||
hf tensor due to the |
nucleus, there are eight EPR absorption lines parallel |
|||
to the |
symmetry axis |
of the |
tensor |
and eight EPR absorption lines |
perpendicular to this axis (Gersmann and Swalen, 1962; Kivelson and Lee, 1964; Albanese and Chasteen, 1978). As seen in Fig. 1, the low-field and high-field absorptions in the spectrum are resolved while in the central region parallel and perpendicular components heavily overlap. The –7/2 parallel and –3/2 perpendicular components designated by arrows correspond to sets of molecules for which the V = O bond lies parallel or perpendicular to 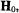 respectively.
respectively.
For the nitroxyl spin-label in Fig. 2, the low-field feature in the EPR
spectrum arises |
from the interaction |
of the |
component |
of the |
tensor |
with the |
projection of the |
nucleus. Since the |
component is |
||
perpendicular to the x,y-plane of the pyrrolinyl ring, molecules for which the spin-label ring lies perpendicularly to 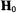 give rise to this feature of the EPR spectrum. Similar geometrical relationships apply also to the high-field absorption feature with
give rise to this feature of the EPR spectrum. Similar geometrical relationships apply also to the high-field absorption feature with  On the other hand, for an X-band spectrometer, the prominent central feature of the frozen solution spectrum of a spin-label arises from heavily overlapping absorption components parallel and perpendicular to the molecular plane. Since the
On the other hand, for an X-band spectrometer, the prominent central feature of the frozen solution spectrum of a spin-label arises from heavily overlapping absorption components parallel and perpendicular to the molecular plane. Since the 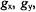 and
and 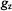 components within the central feature of the EPR absorption spectrum of the nitroxyl group are overlapping at X-band frequencies, this feature is best described, therefore, as arising from a collection of randomly oriented molecules (Wells and Makinen, 1988; Makinen et al., 1998).
components within the central feature of the EPR absorption spectrum of the nitroxyl group are overlapping at X-band frequencies, this feature is best described, therefore, as arising from a collection of randomly oriented molecules (Wells and Makinen, 1988; Makinen et al., 1998).
The principal values of A for the vanadium nucleus and 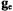 that characterize vanadyl complexes can be extracted from EPR spectra of frozen solutions or polycrystalline samples, as summarized for
that characterize vanadyl complexes can be extracted from EPR spectra of frozen solutions or polycrystalline samples, as summarized for 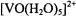 in Table 1. For vanadyl complexes it has been shown that the values of the principal spectroscopic parameters
in Table 1. For vanadyl complexes it has been shown that the values of the principal spectroscopic parameters  and
and  in particular
in particular 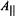 and
and 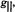 reflect the number and types of equatorial donor-ligand atoms according to chemical element (Chasteen, 1981). Further analysis of this relationship has shown that structural distortion of the complex from square pyramidal to trigonal pyramidal geometry does not alter the contribution of an equatorial donor-atom to
reflect the number and types of equatorial donor-ligand atoms according to chemical element (Chasteen, 1981). Further analysis of this relationship has shown that structural distortion of the complex from square pyramidal to trigonal pyramidal geometry does not alter the contribution of an equatorial donor-atom to 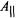 (Cornman et al., 1995, 1997; Smith et al., 2002).
(Cornman et al., 1995, 1997; Smith et al., 2002).
ANGLE-SELECTED ENDOR |
97 |
In the case of the nitroxyl group, in which the unpaired spin density is shared primarily between the N and O atoms (Hayat and Silver, 1973; Davis et al., 1975), the isotropic hf coupling represented by 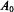 denotes the fraction of spin density associated with the
denotes the fraction of spin density associated with the  atom. This provides a measure of the effective dipolar position of the unpaired spin density associated with the N–O group (Wells and Makinen, 1988; Mustafi et al., 1990a; Makinen et al., 1998). The fraction of spin density associated with the N atom and the effective dipolar position of the unpaired spin may vary slightly according to the dielectric constant of the surrounding medium (Jost and Griffith, 1978). The maximum possible shift amounts to no more than 0.041 Å along the N– O bond between solvents of high and low dielectric constant (Makinen et al., 1998).
atom. This provides a measure of the effective dipolar position of the unpaired spin density associated with the N–O group (Wells and Makinen, 1988; Mustafi et al., 1990a; Makinen et al., 1998). The fraction of spin density associated with the N atom and the effective dipolar position of the unpaired spin may vary slightly according to the dielectric constant of the surrounding medium (Jost and Griffith, 1978). The maximum possible shift amounts to no more than 0.041 Å along the N– O bond between solvents of high and low dielectric constant (Makinen et al., 1998).
2.2Mathematical Formulation of the Physical Basis of ENDOR
ENDOR spectroscopy is performed by “pumping” the electronic transitions of the paramagnetic system under high microwave power and irradiating the system simultaneously with a strong rf field. When the frequency of the rf field is scanned under these conditions and resonance of a nucleus interacting with the unpaired electron is reached, a forbidden transition equivalent to simultaneous electron and nuclear “spin flips” is stimulated, giving rise to increased EPR signal amplitude. Thus, the ENDOR method has its basis in detection of nuclear resonance absorption by observing changes in the intensity of an EPR line.
ENDOR spectroscopy under continuous wave (cw) conditions, as in the studies described here, is optimally carried out with paramagnetic probes with relatively long electronic spin-lattice relaxation times 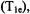 allowing microwave power saturation of the paramagnetic center to be achieved with temperatures ranging from that of liquid helium to that of liquid nitrogen. Since the EPR absorption of
allowing microwave power saturation of the paramagnetic center to be achieved with temperatures ranging from that of liquid helium to that of liquid nitrogen. Since the EPR absorption of 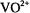 and of nitroxyl spin-labels can be detected at room temperature, subsequent lowering of the temperature readily results in achieving an electronic spin-lattice relaxation time favorable for ENDOR spectroscopy. Also, in our experience, cw ENDOR is optimally detected in systems characterized by narrow EPR absorption features with high peak-to- peak amplitudes. With respect to this latter characteristic, we have found
and of nitroxyl spin-labels can be detected at room temperature, subsequent lowering of the temperature readily results in achieving an electronic spin-lattice relaxation time favorable for ENDOR spectroscopy. Also, in our experience, cw ENDOR is optimally detected in systems characterized by narrow EPR absorption features with high peak-to- peak amplitudes. With respect to this latter characteristic, we have found  and nitroxyl spin-labels to be ideal paramagnetic probes for ENDOR spectroscopy (Makinen and Mustafi, 1995; Makinen et al., 1998).
and nitroxyl spin-labels to be ideal paramagnetic probes for ENDOR spectroscopy (Makinen and Mustafi, 1995; Makinen et al., 1998).
ENDOR spectroscopy provides the most precise method of measuring the strength of electron-nucleus hf interactions. Consequently, its application results in very high resolving power for study of molecular structure. Since an ENDOR spectrum is a nuclear resonance spectrum, one must determine

98 |
DEVKUMAR MUSTAFI AND MARVIN W. MAKINEN |
the total field at the nucleus to calculate the transition frequency. Beginning with the spin Hamiltonian in Eq. (1), it can be shown (Hurst et al., 1985) that the first-order transition frequencies 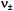 for a nucleus are given by Eq. (2)
for a nucleus are given by Eq. (2)
where |
the are the direction cosines of |
in the molecular axis system, |
|||
is the |
orientation-dependent value of the |
hf coupling, |
is |
the nuclear |
|
Larmor frequency, |
is the electron spin quantum number, and |
represents |
|||
the effective g value defined by the relation  In systems of low g-anisotropy with S = 1/2, the axis of quantization of the unpaired electron spin can be assumed as the direction of the applied magnetic field
In systems of low g-anisotropy with S = 1/2, the axis of quantization of the unpaired electron spin can be assumed as the direction of the applied magnetic field 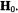 If the applied field is oriented parallel to the principal axis j of the hf tensor A, Eq. (2) simplifies to Eq. (3) where the separation of about
If the applied field is oriented parallel to the principal axis j of the hf tensor A, Eq. (2) simplifies to Eq. (3) where the separation of about  is called the ENDOR shift.
is called the ENDOR shift.
For symmetric separations, the hf coupling is, thus, twice the value of this frequency spacing. Eq. (3) applies to the condition 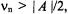 which is characteristic of
which is characteristic of 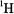 and
and 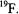 For some nuclei, e. g.,
For some nuclei, e. g., 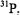 the condition
the condition 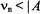
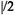 may apply, in which case Eq. (3) becomes
may apply, in which case Eq. (3) becomes 
Within the strong-field approximation, the observed hf coupling A is given by Eq. (4) as a function of r and  where h is the Planck constant, r is
where h is the Planck constant, r is
the modulus of the electron-nucleus position vector r, and |
is the angle |
|||
between |
and r. For |
and nitroxyl spin-label systems, the observed |
||
principal |
hfc components |
and |
correspond, respectively, to the |
|
maximum and minimum ENDOR shifts in the spectrum. The principal hfc
components due to dipole-dipole interaction |
and |
correspond to the |
||||
first term of Eq. (4) for values of |
and 90°, respectively. Under the |
|||||
conditions |
and |
and |
and |
|
|
the traceless dipolar hfc |
components |
can be calculated under the constraint |
|||||
 The signature of the isotropic hfc constants of methyl and the ring hydrogens with respect to that of the nitroxyl nitrogen has been assigned on the basis of TRIPLE spectroscopy (Mustafi and Joela, 1995). These results validate application of the above expressions to assign the magnitude and signs of
The signature of the isotropic hfc constants of methyl and the ring hydrogens with respect to that of the nitroxyl nitrogen has been assigned on the basis of TRIPLE spectroscopy (Mustafi and Joela, 1995). These results validate application of the above expressions to assign the magnitude and signs of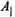 and
and 
The vanadyl ion and nitroxyl spin-labels exhibit low g-anisotropy, as seen in Table 1. Since the pseudo-contact contribution to the isotropic hf
ANGLE-SELECTED ENDOR |
99 |
coupling, represented as the second right-hand term in Eq. (4), is negligible in cases of low g-anisotropy (McConnell and Chestnut, 1958), we have made the approximation that 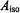 arises entirely from the Fermi contact term. Because the unpaired electron is localized to the metal
arises entirely from the Fermi contact term. Because the unpaired electron is localized to the metal 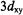 orbital in the
orbital in the 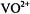 ion (Ballhausen and Gray, 1962) or primarily to the N–O group in spin-labels (Hayat and Silver, 1973; Mustafi et al., 1991), the transfer of unpaired spin density to other atoms in the system is small. This aspect is of particular importance, for it ensures valid application of the point-dipole approximation, allowing structure determination with high precision.
ion (Ballhausen and Gray, 1962) or primarily to the N–O group in spin-labels (Hayat and Silver, 1973; Mustafi et al., 1991), the transfer of unpaired spin density to other atoms in the system is small. This aspect is of particular importance, for it ensures valid application of the point-dipole approximation, allowing structure determination with high precision.
Snetsinger et al. (1992) have shown that attributing the observed hf coupling of an unpaired electron in a metal 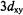 orbital entirely to the dipoledipole interaction with a nuclear spin of I = 1/2 over a distance of 2.09 Å results in a calculated separation of 2.15 Å. This result applies directly to the
orbital entirely to the dipoledipole interaction with a nuclear spin of I = 1/2 over a distance of 2.09 Å results in a calculated separation of 2.15 Å. This result applies directly to the  ion, and the difference of 0.06 Å can be considered essentially inconsequential. Since the contribution of the isotropic hfc is less for larger electron-nucleus distances, the error will be correspondingly smaller. The shortest vanadium-proton distance that we have measured is of the order 2.6 Å, corresponding to the protons of solvent molecules in the inner coordination sphere of the
ion, and the difference of 0.06 Å can be considered essentially inconsequential. Since the contribution of the isotropic hfc is less for larger electron-nucleus distances, the error will be correspondingly smaller. The shortest vanadium-proton distance that we have measured is of the order 2.6 Å, corresponding to the protons of solvent molecules in the inner coordination sphere of the  ion (Mustafi and Makinen, 1988).
ion (Mustafi and Makinen, 1988).
In the case of nitroxyl spin-labels, the important electron-nucleus distances for structure analysis correspond invariably to nuclei not immediately attached to the pyrrolinyl ring. However, even in the case of hydrogens attached to ring carbon atoms, the error remains negligible. The electron–nucleus separation measured by ENDOR for the vinyl proton in 2,2,5,5-tetramethyl-1oxypyrroline-3-carboxamide was 3.78 ± 0.01 Å (Mustafi et al., 1990c, 1991). The distance based on X-ray coordinates (Turley and Boer, 1972) yields 3.79 Å. In general, we have shown that errors associated with the point-dipole and strong-field approximations are less than 5% for  Å in both types of paramagnetic systems. Furthermore, assignment of the effective dipolar position of the unpaired spin of the N–O group on the basis of ENDOR-determined electron-nucleus distances has been made to within ± 0.04 Å (Mustafi et al., 1991). We, thus, conclude that errors originating from the point-dipole and strong-field approximations are negligible (Makinen and Mustafi, 1995; Makinen et al., 1998).
Å in both types of paramagnetic systems. Furthermore, assignment of the effective dipolar position of the unpaired spin of the N–O group on the basis of ENDOR-determined electron-nucleus distances has been made to within ± 0.04 Å (Mustafi et al., 1991). We, thus, conclude that errors originating from the point-dipole and strong-field approximations are negligible (Makinen and Mustafi, 1995; Makinen et al., 1998).
2.3Characteristics of ENDOR Spectra at Selected Molecular Orientations
To determine structural information by ENDOR, one must (i) identify the surrounding nuclei contributing to hf interactions and (ii) determine the dependence of ENDOR spectra on the settings of the external magnetic field with respect to magnetic axes in the molecule. For  and
and