

Biomedical EPR Part-B Methodology Instrumentation and Dynamics - Sandra R. Eaton
.pdf120 |
DEVKUMAR MUSTAFI AND MARVIN W. MAKINEN |
Figure 15. Stereo diagram of MD-averaged spin-labeled methyl L-tryptophanate in high and low dielectric solvents. The spin-label moieties for each conformation, including the carbonyl group adjacent to the spin-label ring, were superimposed to highlight the change in the orientation of the indole side chain. The molecule rendered black represents the conformer of higher population found in high dielectric solvents 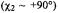 while that rendered gray corresponds to the conformer of higher population in low dielectric solvents
while that rendered gray corresponds to the conformer of higher population in low dielectric solvents  See Table 5 for definition of
See Table 5 for definition of  Reprinted from Van Zele et al. (2001) with permission.
Reprinted from Van Zele et al. (2001) with permission.
Since bond lengths of nonhydrogen atoms and valence angles are often not fixed in MD simulations of small molecules, simulation of the dynamical motion of spin-labeled molecules constrained by ENDOR data with application of the forcefield parameters for the spin-label moiety derived by Van Zele et al. (2001) represents a significantly improved means for structural analysis compared to the rigid body approach with fixed molecular fragments that has been hitherto necessary (Makinen, 1998; Makinen et al., 1998). On this basis, structure analysis would be comparable to present day applications of simulated annealing calculations of protein and polypeptide structure in which nuclear Overhauser distances are incorporated as restraints in NMR studies (Herrmann et al., 2002). ENDOR distance constraints, while fewer in number, are, however, of significantly higher precision than the inter-nuclear distances determined by NMR (Zhao and Jardetzky, 1994). Further exploration of this approach in ENDOR structural analysis should be pursued and applied to macromolecular systems since it offers the most accurate and precise method of defining local structural details, for instance, those in active sites of enzyme reaction intermediates that are likely to be of catalytic significance.
3.2.2Structured solvent molecules hydrogen-bonded to spin-labeled
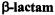 antibiotics
antibiotics
The penam and cepham fused ring structures of first generation 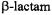 antibiotics belonging, respectively, to 6-aminopenicillanic acid and 7- aminocephalosporanic acid are illustrated in Fig. 16. The free amine forms of the
antibiotics belonging, respectively, to 6-aminopenicillanic acid and 7- aminocephalosporanic acid are illustrated in Fig. 16. The free amine forms of the 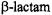 antibiotics exhibit no inhibitory action against pathogenic bacteria (Bush et al., 1995; Massova and Mobashery, 1998). However,
antibiotics exhibit no inhibitory action against pathogenic bacteria (Bush et al., 1995; Massova and Mobashery, 1998). However,
ANGLE-SELECTED ENDOR |
121 |
derivatization of the amine group confers antimicrobial activity. The spinlabels IV in Fig. 10, used to derivatize the amine group of penicillin and cephalosporin in our ENDOR studies, are sterically and structurally analogous to a variety of acylamido groups found in clinically useful antibiotics.
Figure 16. Chemical bonding structures of 6-aminopenicillanic acid (left) and 7- aminocephalosporanic acid (right), illustrating penam and cepham fused ring structures, respectively, of first generation  antibiotics.
antibiotics.
We have observed through ENDOR studies that the –NH– group of the acyl-amido linkage of free, spin-labeled antibiotics in solution exhibits a pronounced tendency for hydrogen-bonding to solvent molecules (Mustafi and Makinen, 1995; Mustafi et al., 1997). In Fig. 12 the two narrow line pairs belonging to the OH group of the hydrogen-bonded methanol molecule appearing upon addition of only one equivalent of methanol in an anhydrous aprotic solvent indicate that methanol forms a tightly bound adduct with the SLCEP molecule. Since the parallel and perpendicular resonances of  appear in the spectrum for setting B, the hydroxyl proton must lie in the plane of the spin-label according to the requirements of angle selection, as summarized in Fig. 4. A search for sites on the SLCEP molecule capable of forming a tightly bound adduct with the OH group of the methanol molecule such that the electron-proton distance of 5.66 ± 0.03 Å is satisfied and that the proton lies in the plane of the spin-label leaves only the acylamido –NH– group as a possible candidate for hydrogen-bonding. Moreover, by searching sterically accessible conformational space, we determined that the dipolar electron-proton distance to
appear in the spectrum for setting B, the hydroxyl proton must lie in the plane of the spin-label according to the requirements of angle selection, as summarized in Fig. 4. A search for sites on the SLCEP molecule capable of forming a tightly bound adduct with the OH group of the methanol molecule such that the electron-proton distance of 5.66 ± 0.03 Å is satisfied and that the proton lies in the plane of the spin-label leaves only the acylamido –NH– group as a possible candidate for hydrogen-bonding. Moreover, by searching sterically accessible conformational space, we determined that the dipolar electron-proton distance to 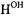 is compatible with the OH group hydrogenbonded to the acylamido NH group only on the endo or concave surface of the
is compatible with the OH group hydrogenbonded to the acylamido NH group only on the endo or concave surface of the  group (Mustafi et al., 1997). In Fig. 14 the dotted surface representing the solvent accessible surface (Lee and Richards, 1971) shows that the OH group of the methanol molecule positioned according to ENDOR structural constraints is sterically accommodated. Water molecules are found hydrogen-bonded to the acylamido –NH– group on the endo surface in the X-ray structures of cephaloglycin (Sweet and Dahl, 1970) and amoxicillin (Boles et al., 1978). By ENDOR we have similarly assigned a
group (Mustafi et al., 1997). In Fig. 14 the dotted surface representing the solvent accessible surface (Lee and Richards, 1971) shows that the OH group of the methanol molecule positioned according to ENDOR structural constraints is sterically accommodated. Water molecules are found hydrogen-bonded to the acylamido –NH– group on the endo surface in the X-ray structures of cephaloglycin (Sweet and Dahl, 1970) and amoxicillin (Boles et al., 1978). By ENDOR we have similarly assigned a

122 DEVKUMAR MUSTAFI AND MARVIN W. MAKINEN
hydrogen-bonded solvent molecule on the endo surface of SLPEN (Mustafi and Makinen, 1995).
The 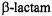 C–N bond of antibiotics free in solution is cleaved through solvolytic reactions and is the specific point of attack by
C–N bond of antibiotics free in solution is cleaved through solvolytic reactions and is the specific point of attack by 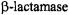 enzymes. It is of particular interest to compare the known stereochemistry of solvolytic and enzyme-catalyzed reactions. The steric approach of the hydrolytic solvent molecule has not been resolved for free
enzymes. It is of particular interest to compare the known stereochemistry of solvolytic and enzyme-catalyzed reactions. The steric approach of the hydrolytic solvent molecule has not been resolved for free 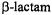 antibiotics in solution and remains conjectural. Possible pathways for nucleophilic attack of the
antibiotics in solution and remains conjectural. Possible pathways for nucleophilic attack of the  carbonyl carbon are illustrated in Fig. 17. Attack from the exo or concave surface is stereoelectronically forbidden while nucleophilic attack from the endo or concave surface is stereoelectronically allowed (Deslongchamps, 1983; Benner, 1988). The hydrogen-bonded solvent molecules located on the endo or concave surface of
carbonyl carbon are illustrated in Fig. 17. Attack from the exo or concave surface is stereoelectronically forbidden while nucleophilic attack from the endo or concave surface is stereoelectronically allowed (Deslongchamps, 1983; Benner, 1988). The hydrogen-bonded solvent molecules located on the endo or concave surface of 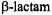 antibiotics defined by X-ray crystallographic data have been ignored in proposals of the mechanistic pathway for solvolysis (Page, 1987). Consequently it has been thought that nucleophilic attack can occur only on the exo or convex surface of the
antibiotics defined by X-ray crystallographic data have been ignored in proposals of the mechanistic pathway for solvolysis (Page, 1987). Consequently it has been thought that nucleophilic attack can occur only on the exo or convex surface of the  ring because it was assumed that the endo surface could not sterically accommodate a solvent molecule. However, ENDOR identification of hydrogen-bonded solvent only on the endo surface for both SLCEP and SLPEN indicates that the solvolytic reaction of free
ring because it was assumed that the endo surface could not sterically accommodate a solvent molecule. However, ENDOR identification of hydrogen-bonded solvent only on the endo surface for both SLCEP and SLPEN indicates that the solvolytic reaction of free  antibiotics in solution could proceed via endo nucleophilic attack, consistent with stereoelectronic rules (Mustafi and Makinen, 1995; Mustafi et al., 1997).
antibiotics in solution could proceed via endo nucleophilic attack, consistent with stereoelectronic rules (Mustafi and Makinen, 1995; Mustafi et al., 1997).
Figure 17. Schematic drawing of the molecular structure of  antibiotics to illustrate the exo or convex surface (from top side, sterically-preferred path, but stereoelectronically forbidden path for nucleophilic attack), and endo or concave surface (from bottom side, sterically more restricted, but stereoelectronically allowed path for nucleophilic attack). Note that in the case of endo attack, the path of the incoming nucleophile is antiperiplanar to the lone pair on the
antibiotics to illustrate the exo or convex surface (from top side, sterically-preferred path, but stereoelectronically forbidden path for nucleophilic attack), and endo or concave surface (from bottom side, sterically more restricted, but stereoelectronically allowed path for nucleophilic attack). Note that in the case of endo attack, the path of the incoming nucleophile is antiperiplanar to the lone pair on the  nitrogen atom, as required for stereoelectronically allowed hydrolysis. On the other hand, the exo approach of an electron-rich nucleophile undergoes repulsive interactions with the lone pair orbital. Protonation of the
nitrogen atom, as required for stereoelectronically allowed hydrolysis. On the other hand, the exo approach of an electron-rich nucleophile undergoes repulsive interactions with the lone pair orbital. Protonation of the  nitrogen prior to nucleophilic attack renders exo and endo surfaces stereoelectronically equivalent.
nitrogen prior to nucleophilic attack renders exo and endo surfaces stereoelectronically equivalent.
Interestingly, in TEM-1 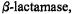 and, therefore, presumably in all other homologous class A
and, therefore, presumably in all other homologous class A  the active site Ser70 side chain and the deacylating water molecule approach the carbonyl carbon atom via the exo surface of the
the active site Ser70 side chain and the deacylating water molecule approach the carbonyl carbon atom via the exo surface of the  substrate (Strynadka et al., 1992). Protonation of
substrate (Strynadka et al., 1992). Protonation of
ANGLE-SELECTED ENDOR |
123 |
the 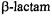 nitrogen prior to nucleophilic attack renders exo and endo surfaces stereoelectronically equivalent (Mustafi and Makinen, 1995) and protonation of the
nitrogen prior to nucleophilic attack renders exo and endo surfaces stereoelectronically equivalent (Mustafi and Makinen, 1995) and protonation of the 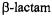 nitrogen prior to exo nucleophilic attack is energetically favored (Atanasov et al., 2000). In class C
nitrogen prior to exo nucleophilic attack is energetically favored (Atanasov et al., 2000). In class C 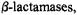 however, the approach of the hydrolytic water in the acylenzyme reaction intermediate is thought to proceed from the endo surface (Massova and Mobashery, 1998). Therefore, the hydrolytic mechanism of class C
however, the approach of the hydrolytic water in the acylenzyme reaction intermediate is thought to proceed from the endo surface (Massova and Mobashery, 1998). Therefore, the hydrolytic mechanism of class C 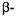
 is likely to differ from that associated with class A enzymes. Further details of the mechanistic pathway of
is likely to differ from that associated with class A enzymes. Further details of the mechanistic pathway of 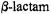 hydrolysis in class C enzymes have yet to be defined.
hydrolysis in class C enzymes have yet to be defined.
3.3ENDOR Detection of the Hydrolytic Water in a
Spin-labeled Acylenzyme Reaction Intermediate of TEM-1 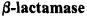
3.3.1Cryokinetic isolation of enzyme reaction intermediates
Much of our understanding of the structural basis of enzyme catalytic action is derived from X-ray analysis of enzyme-inhibitor complexes. Such complexes mimic in part enzyme reaction intermediates, but they are inherently chemically stable because of non-productive spatial relationships. On the other hand, true enzyme reaction intermediates are chemically labile because of catalytically productive spatial relationships. While the active site interactions induced by an inhibitor that is structurally similar to a substrate may overlap in part with those required for catalysis, the two different sets of enzyme-ligand interactions cannot be identical. Thus, in any enzymeinhibitor complex at least one critical structural interaction will be absent that is required for catalysis. Nonetheless, mechanisms of action of enzymes have been generally derived through modeling based on active site spatial relationships observed in enzyme-inhibitor complexes.
There are salient instances in which mechanistic conclusions on the basis of only X-ray structure analysis have proven to be misleading. The recent identification of a covalent reaction intermediate of hen egg-white lysozyme (Vocadlo et al., 2001), in contrast to the general acid catalyzed mechanism involving no covalent interactions between the substrate and enzyme proposed on the basis of X-ray defined inhibitor complexes (Phillips, 1967; Stryer, 1988), serves as a prominent reminder. In addition, EPR and ENDOR characterization of a covalent (mixed anhydride) acylenzyme intermediate of carboxypeptidase A (Makinen et al., 1979; Kuo et al., 1983; Mustafi and Makinen, 1994) stands in contradistinction to the general base catalyzed mechanism favored through X-ray studies (Christianson and Lipscomb, 1986, 1989) since an enzyme-product complex was shown not to account for
124 DEVKUMAR MUSTAFI AND MARVIN W. MAKINEN
the spectroscopic results. Furthermore, nucleophile trapping experiments have demonstrated a mixed anhydride linkage for both esterolytic and proteolytic substrates of carboxypeptidase A (Sander and Witzel, 1985, 1986). These differences highlight the importance of identifying true reaction intermediates for structural studies. For these reasons, characterization of true, catalytically competent enzyme reaction intermediates should always be the preferred approach to define the structural basis of enzyme action.
We have found application of cryoenzymologic methods in combination with angle-selected ENDOR to be highly suitable for defining threedimensional active site structure in catalytically competent intermediates of enzyme-catalyzed reactions. Cryoenzymology entails establishing, by experiment, conditions in the 0° C to –90° C range under which transient intermediates of enzyme-catalyzed reactions can be more readily detected and temporally resolved than at room temperature because of their intrinsically longer half-lives at low temperatures (Douzou, 1977; Makinen and Fink, 1977; Fink and Geeves, 1979; Fink and Cartwright, 1981). Cryokinetic isolation of an enzyme reaction intermediate is carried out in fluid, cosolvent mixtures at low temperatures which form glasses upon freezing. In contrast to frozen water or ice, which is crystalline, glasses are by definition isotropic in structure, and, therefore, are not expected to be associated with perturbation of protein conformation. In our studies ENDOR must be carried out on solid state systems (to detect the dipolar hf interactions through which structural relationships are defined) and at low temperatures (to saturate relaxation processes with microwave power that govern detection of resonance absorption). Consequently, preparation of enzyme reaction intermediates in cryosolvent mixtures followed by freezequenching of the reaction mixture is ideal for ENDOR spectroscopy.
The basis of cryoenzymology is very similar in principle to cryocrystallography. In cryo-crystallography, cryoprotectant cosolvents and flash-cooling methods are employed, particularly when synchrotron radiation is used to collect diffraction data. The protein crystal is equilibrated with an aqueous, cryoprotectant, cosolvent mixture followed by flashfreezing in liquid nitrogen (Garman and Schneider, 1997; Rodgers, 1997). The cryoprotectant cosolvents employed are generally polyol substances such as glycerol, ethylene glycol, or 2-methyl-2,4-pentanediol which can be also used in cryoenzymology (Douzou, 1977). Because of the high aqueous solvent content of protein crystals (Matthews, 1968), it is believed that freezing of the cryoprotectant cosolvent mixture results in glass formation within the solvent channels of the crystal (Rodgers, 1997). This is essentially not different from freezing of enzyme complexes and enzyme reaction intermediates in cryosolvent mixtures for ENDOR. Since 1995, no less than
ANGLE-SELECTED ENDOR |
125 |
40% of X-ray determined protein structures have been carried out by application of cryo-crystallographic methods with data collection at 100 K or lower (Garman and Schneider, 1997). Teng and Moffat (1998) have pointed out that factors that can potentially perturb protein structure using cryocooled crystals, such as rate of crystal cooling, choice of cryosolvent, change in crystal mosaic spread, and binding of cryosolvent to the protein in the crystal, to name a few, have not been evaluated in detail. On the other hand, we have not observed deleterious effects of cryosolvent mixtures on protein structure upon freezing of cryosolvent mixtures, provided appropriate precautions are taken during introduction of the organic cosolvent to avoid protein denaturation. Since denaturation of proteins is subject to high activation barriers, this problem can be avoided by coordinating lowering of the temperature with introduction of the organic cosolvent in small aliquots (Douzou, 1977; Fink and Geeves, 1979).
There are important advantages to structurally characterize intermediates of enzyme-catalyzed reactions through a combined approach of cryoenzymology applied in conjunction with ENDOR spectroscopy:
(i) It is often possible, depending on the forward and backward rate constants of the enzyme-catalyzed reaction, to achieve near-stoichiometric conversion of free enzyme to reaction intermediate. This cannot be as easily achieved through use of rapid-flow, freeze-quench methods (Makinen and Fink, 1977).
(ii) The half-life of a species entering a unimolecular reaction (generally typical of each sequential step in a one-substrate, enzyme-catalyzed reaction after formation of the Michaelis complex) is given by the relationship 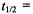
 where k is the rate constant governing the step. Thus, the half-life can be made significantly longer than the mixing time by achieving a small enough value of k at low temperatures. This condition ensures that the concentrations of breakdown products are negligible. Furthermore, for unimolecular reactions with enzyme in excess, it is straightforward to identify conditions under which the substrate is virtually totally bound, i.e., saturated by the enzyme, in forming a reaction intermediate. This is especially important with use of substrates that are synthetically designed to serve as the paramagnetic structural probe. Thus, overlapping spectroscopic effects due to free and bound substrate or product can be avoided.
where k is the rate constant governing the step. Thus, the half-life can be made significantly longer than the mixing time by achieving a small enough value of k at low temperatures. This condition ensures that the concentrations of breakdown products are negligible. Furthermore, for unimolecular reactions with enzyme in excess, it is straightforward to identify conditions under which the substrate is virtually totally bound, i.e., saturated by the enzyme, in forming a reaction intermediate. This is especially important with use of substrates that are synthetically designed to serve as the paramagnetic structural probe. Thus, overlapping spectroscopic effects due to free and bound substrate or product can be avoided.
(iii) Cryokinetic characterization of the enzyme-catalyzed reaction under conditions of enzyme in excess allows direct application of the reaction conditions for preparing reaction intermediates for ENDOR studies. Thus, there is no question whether the conditions employed for kinetic characterization are relevant to those employed for isolation of reaction intermediates for structure determination.

126 |
DEVKUMAR MUSTAFI AND MARVIN W. MAKINEN |
Furthermore, cryostabilized reaction intermediates do not represent “trapped, low-energy structures.” For instance, we have demonstrated by ENDOR spectroscopy that the eclipsed conformation of the substrate moiety in the spin-labeled tryptophanyl acylenzyme intermediate of 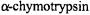 stabilized in cryosolvents (Wells et al., 1994) is identical to that detected in a complex of the enzyme formed at room temperature with a spin-labeled transition-state analog (Jiang et al., 1998; Makinen, 1998). Such eclipsed dihedral angle relationships are indicative of conformations of high potential energy and are not characteristic of the ground state species. When cryostabilized enzyme reaction intermediates are formed according to kinetic criteria, they represent catalytically competent species even though they have been generated in cosolvent mixtures at low temperatures.
stabilized in cryosolvents (Wells et al., 1994) is identical to that detected in a complex of the enzyme formed at room temperature with a spin-labeled transition-state analog (Jiang et al., 1998; Makinen, 1998). Such eclipsed dihedral angle relationships are indicative of conformations of high potential energy and are not characteristic of the ground state species. When cryostabilized enzyme reaction intermediates are formed according to kinetic criteria, they represent catalytically competent species even though they have been generated in cosolvent mixtures at low temperatures.
3.3.2The catalytic role of sequestered water in the active site of TEM-1 
Resistance to |
antibiotics has become |
a serious public health |
threat because of the widespread distribution of |
in pathogenic |
|
bacteria and because of their continual ability to produce mutant enzymes to avoid drug action (Neu, 1992). Therefore, it is important to understand not only structural determinants of antibiotic substrate recognition among the various classes of  but also structural differences that underlie their mechanisms of action. The antimicrobial action of penicillin and cephalosporin antibiotics is due to the reactivity of the four-membered
but also structural differences that underlie their mechanisms of action. The antimicrobial action of penicillin and cephalosporin antibiotics is due to the reactivity of the four-membered 
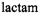 ring (cf., Fig. 16), on which basis inhibition of cell wall synthesis occurs in bacteria. The DD-transpeptidase enzyme that generates the peptidoglycan polymer of the bacterial cell wall forms an acylenzyme with
ring (cf., Fig. 16), on which basis inhibition of cell wall synthesis occurs in bacteria. The DD-transpeptidase enzyme that generates the peptidoglycan polymer of the bacterial cell wall forms an acylenzyme with
antibiotics with a half-life |
12 hours. This results in cell death. |
||
Resistance to |
antibiotics by |
pathogenic bacteria has |
developed |
through evolving |
highly efficient |
enzymes |
that are |
evolutionarily derived from the DD-transpeptidase and are secreted into the periplasmic space of the bacterium. Thus, they are exposed to the antibiotic before it can penetrate to reach the membrane-bound DD-transpeptidase enzyme. Thus, while  antibiotics are mechanism-based inactivators of the target DD-transpeptidase enzyme, they are catalytically specific substrates of the
antibiotics are mechanism-based inactivators of the target DD-transpeptidase enzyme, they are catalytically specific substrates of the 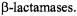
The interaction of the |
of the serine hydrolase variety with |
|
antibiotics is |
schematically |
illustrated in Fig, 18. Although the |
structures of a number |
of |
have been solved to high resolution |
(Jelsch et al., 1993; Knox, 1995; Paetzel et al., 2000), there is relatively little agreement about the chemical roles of active site residues except for that of the active site nucleophilic serine residue. The side chain attacks the

ANGLE-SELECTED ENDOR |
127 |
carbonyl carbon of the 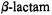 group. In class A
group. In class A  of which the TEM-1 enzyme is a prominent example, it is anticipated, in analogy to serine proteases, that a general-base participates in catalysis in the active site by abstracting a proton from the side chain of Ser70 during formation of the transient tetrahedral adduct. However, the identity of the residue serving this function and governing catalytic cleavage with a
of which the TEM-1 enzyme is a prominent example, it is anticipated, in analogy to serine proteases, that a general-base participates in catalysis in the active site by abstracting a proton from the side chain of Ser70 during formation of the transient tetrahedral adduct. However, the identity of the residue serving this function and governing catalytic cleavage with a 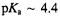 has remained conjectural.
has remained conjectural.
Figure 18. Schematic illustration of the hydrolytic reaction underlying the interaction of penicillin-recognizing enzymes of the serine hydrolase type with 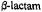 antibiotics. Formation of the Michaelis complex (ES) is followed by acylation of the nuclephilic serine side chain in the active site. The acylenzyme (EY) undergoes rate-limiting hydrolysis leading to destruction of the antibiotic potency of the
antibiotics. Formation of the Michaelis complex (ES) is followed by acylation of the nuclephilic serine side chain in the active site. The acylenzyme (EY) undergoes rate-limiting hydrolysis leading to destruction of the antibiotic potency of the 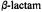 compound. For the DD-transpeptidase representing the target enzyme of the antibiotic,
compound. For the DD-transpeptidase representing the target enzyme of the antibiotic,  is very low,
is very low,  while for
while for 
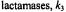 can be of the order of
can be of the order of 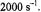
It has been suggested through X-ray studies of a deacylation defective mutant of TEM-1 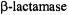 that Lys73, while hydrogen-bonded to Ser70 in the free enzyme, acts as a neutral base in the Michaelis complex and
that Lys73, while hydrogen-bonded to Ser70 in the free enzyme, acts as a neutral base in the Michaelis complex and
functions as a proton acceptor |
(Strynadka et al., 1992). However, NMR |
|
titration studies of TEM-1 |
biosynthetically enriched with |
|
lysine show that the |
of Lys73 is >10, whereby Damblon and coworkers |
|
(1996) suggest by elimination that the only residue left in the active site to function as a general-base and to account for the 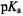 is Glu166. By cryokinetic isolation of a true catalytically competent acylenzyme reaction intermediate, we were able to identify by ENDOR the hydrolytic water molecule hydrogen-bonded to the side chain of Glu166, confirming its role as the general base catalyst in the active site (Mustafi et al., 2001). We were also able to demonstrate that the proposed functional role of Lys73 as a proton acceptor (Strynadka et al., 1992) was not structurally compatible with the active site geometry of the acylenzyme intermediate.
is Glu166. By cryokinetic isolation of a true catalytically competent acylenzyme reaction intermediate, we were able to identify by ENDOR the hydrolytic water molecule hydrogen-bonded to the side chain of Glu166, confirming its role as the general base catalyst in the active site (Mustafi et al., 2001). We were also able to demonstrate that the proposed functional role of Lys73 as a proton acceptor (Strynadka et al., 1992) was not structurally compatible with the active site geometry of the acylenzyme intermediate.
Fig. 19 illustrates ENDOR spectra of chemically modified amino acid side chains that served as probes of active site structure in the deuterated enzyme. The resonance feature in the upper set of spectra is assigned to the  group of acetyl-Tyr105 through its disappearance upon reaction of the enzyme with
group of acetyl-Tyr105 through its disappearance upon reaction of the enzyme with 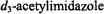 (the next nearest tyrosinyl residue is > 17 Å, rendering it undetectable). Similarly, the lower set of spectra in Fig. 19 belongs to the Glu240Cys mutant, in which the mutant cysteinyl side chain
(the next nearest tyrosinyl residue is > 17 Å, rendering it undetectable). Similarly, the lower set of spectra in Fig. 19 belongs to the Glu240Cys mutant, in which the mutant cysteinyl side chain
has an |
attached |
group. The decrease in intensity for the deuterated |
|
analog |
identifies |
the resonance feature belonging to the |
group. |

128 |
DEVKUMAR MUSTAFI AND MARVIN W. MAKINEN |
Similar results were also obtained for the acyl-enzyme of the Met272-Cys mutant.
Figure 19. Comparison of proton ENDOR spectra of spin-labeled acylenzyme reaction intermediates formed with acetylated (wild type) TEM-1 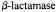 (upper set of spectra, labeled a) and with the Glu240Cys mutant in which the Cys240 side chain has been modified with methylmethanethiolsulfonate (MMTS) (lower set of spectra, labeled b). In each case, enzyme biosynthetically enriched with deuterium (88-90%
(upper set of spectra, labeled a) and with the Glu240Cys mutant in which the Cys240 side chain has been modified with methylmethanethiolsulfonate (MMTS) (lower set of spectra, labeled b). In each case, enzyme biosynthetically enriched with deuterium (88-90% 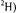 was used. The spectrum for each acylenzyme intermediate reacted with acetylimidazole, correspondingly, MMTS (top spectrum in each set) is compared to the spectrum obtained for the enzyme reacted with
was used. The spectrum for each acylenzyme intermediate reacted with acetylimidazole, correspondingly, MMTS (top spectrum in each set) is compared to the spectrum obtained for the enzyme reacted with 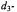
 or
or 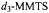 (bottom spectrum in each set). The resonance feature specific for each chemically modified side chain, highlighted by an arrow, is absent in the spectrum of the deuterated analog. Reprinted from Mustafi et al. (2001) with permission.
(bottom spectrum in each set). The resonance feature specific for each chemically modified side chain, highlighted by an arrow, is absent in the spectrum of the deuterated analog. Reprinted from Mustafi et al. (2001) with permission.
Fig. 20 illustrates the spin-labeled penicilloyl moiety in the active site of TEM-1 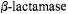 and its structural relationships to the three side chains of active site residues that have been modified for use as ENDOR probes. Their respective electronnucleus distances are indicated in the figure. In separate experiments the resonance of the H(6) proton on the penicilloyl moiety of the acylenzyme yielded an electron-nucleus distance of 6.54 ± 0.10 Å (Mustafi et al., 2001). This constraint, together with the distance constraints to chemically modified amino acid side chains in the active site, allowed virtually no additional degrees of freedom to accommodate the substrate in its catalytically competent conformation due to the hard-sphere, van der Waals radii of other active site residues and the fixed geometry of the ester bond between the acyl moiety of the substrate and the side chain of Ser70.
and its structural relationships to the three side chains of active site residues that have been modified for use as ENDOR probes. Their respective electronnucleus distances are indicated in the figure. In separate experiments the resonance of the H(6) proton on the penicilloyl moiety of the acylenzyme yielded an electron-nucleus distance of 6.54 ± 0.10 Å (Mustafi et al., 2001). This constraint, together with the distance constraints to chemically modified amino acid side chains in the active site, allowed virtually no additional degrees of freedom to accommodate the substrate in its catalytically competent conformation due to the hard-sphere, van der Waals radii of other active site residues and the fixed geometry of the ester bond between the acyl moiety of the substrate and the side chain of Ser70.
While the resonances described above required the use of heavily deuterated enzyme (Sosa-Peinado et al., 2000) in perdeuterated solvent, identification
ANGLE-SELECTED ENDOR |
129 |
of sequestered solvent molecules in the active site required protiated solvent. Fig. 21 compares ENDOR spectra of deuterium enriched spin-labeled reaction intermediates of the wild type and Glu166Asn enzymes in protiated solvent. The resonance features, labeled H’ and H”, cannot be attributed to bulk solvent and, therefore, must arise from solvent exchangeable protons of amino acid residues or solvent molecules sequestered in the protein. Of critical importance here is the comparison of the wild type spectra to those of the Glu166Asn mutant enzyme. This mutant lacks the Glu-166 side chain and, therefore, is unable to catalyze deacylation. The H” resonance in Fig. 21 is common to both the wild type and Glu166Asn mutant enzymes, yielding an electron-nucleus distance of 5.61 ± 0.10 Å radius. This resonance was assigned to the 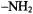 group of Asnl32, the only residue with solvent exchangeable hydrogens in the active site satisfying both the distance constraint and the angle selection requirement of lying in the plane of the spin-label (Mustafi et al., 2001).
group of Asnl32, the only residue with solvent exchangeable hydrogens in the active site satisfying both the distance constraint and the angle selection requirement of lying in the plane of the spin-label (Mustafi et al., 2001).
Figure 20. Stereo view of the active site of the acylenzyme of TEM-1  formed with spin-labeled penicillin (SLPEN). The conformation of the substrate is constrained by the ENDOR-determined electron-H(6) distance in the spin-labeled penicilloyl moiety. Electronproton distances from the unpaired electron of the nitroxyl group to the methyl group of acetyl-Tyr105 (7.59 Å) and to the thiomethoxy groups attached to the mutant cysteinyl side chains in the Glu240Cys (6.57 Å) and Met272Cys (6.91 Å) enzymes are also indicated. The dotted surface represents the calculated Lee-Richards solvent accessible surface (Lee and Richards, 1971) of the active site. Reprinted from Mustafi et al. (2001) with permission.
formed with spin-labeled penicillin (SLPEN). The conformation of the substrate is constrained by the ENDOR-determined electron-H(6) distance in the spin-labeled penicilloyl moiety. Electronproton distances from the unpaired electron of the nitroxyl group to the methyl group of acetyl-Tyr105 (7.59 Å) and to the thiomethoxy groups attached to the mutant cysteinyl side chains in the Glu240Cys (6.57 Å) and Met272Cys (6.91 Å) enzymes are also indicated. The dotted surface represents the calculated Lee-Richards solvent accessible surface (Lee and Richards, 1971) of the active site. Reprinted from Mustafi et al. (2001) with permission.
On the other hand, the H’ feature yielding a 6.65 ± 0.10 Å electronnucleus distance is not observed for the acylenzyme of the Glu166Asn mutant. It must arise, therefore, in a region of the wild type enzyme that differs from the mutant, i.e., the immediate environment of the Glu166 side chain. Since we found no other residues with exchangeable protons satisfying the ENDOR spectroscopic constraints, we ascribed the H’ resonance to sequestered water (Mustafi et al., 2001). To identify the