

Astruc D. - Modern arene chemistry (2002)(en)
.pdf6.3 Tetraethynylethene Dimers 203
Scheme 5. Synthesis of TEE dimers by selective desilylation-oxidative coupling methodology [29]. i) K2CO3, MeOH, THF; ii) anhydrous Cu(OAc)2, pyridine, air (for 19) or CuCl, TMEDA, CH2Cl2, air (for 20– 24). TMEDA ¼ N,N,N0,N0-tetramethylethylenediamine.
The electronic properties of these dimers were studied by UV/vis spectrophotometry. The absorption spectra of the anilino-substituted compounds 20, 21, and 24 display broad absorption shoulders at low energy (lmax in the range 550–580 nm), with end-absorptions extending to 600 nm and beyond. These bands were assigned to charge-transfer (CT) transitions; this was confirmed by protonation with conc. aqueous HCl of the electron-donating anilino groups, which resulted in complete loss of the CT transition [29d]. From these studies it was inferred that the central conjugated C20 core in the TEE dimers, with its 16 C(sp) atoms, is a strong electron-acceptor and that the electron-accepting properties are only weakly enhanced by the two 4-nitrophenyl substituents in the donor–acceptor push–pull system 24. Indeed, this strong acceptor property is not surprising, taking into account the ability of nitrophenyl-substituted TEEs to convey a negative charge from the nitrophenyl group into the carbon framework, as described in the previous paragraph. Moreover, absorption spectra of anilino-substituted TEE monomers reveal a CT transition that is lost upon protonation [12b, 22]. For both the TEE monomers and dimers, this quenching proved to be reversible: treatment of the protonated forms with NaOH regenerated the neutral species, and the UV/vis spectra became virtually identical to those measured before acid treatment. In addition, the first reduction of 20 (Eo ¼ 1:61 V versus Fc/Fcþ in CH2Cl2 [29d]) is cathodically shifted relative to that of 19 (Eo ¼ 1:52 V versus Fc/Fcþ in THF [29b]) as a result of electron donation from the anilino groups into the carbon core of 20. The anilino groups of 20 are oxidized in two reversible two-electron steps at þ0.37 and þ0.54 V.
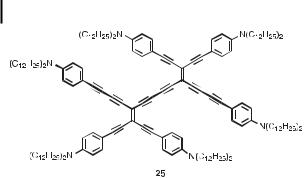
Further extension of the chromophores in 25, by two anilinoethynyl groups (Figure 5), leads to a significant bathochromic shift of the longest-wavelength absorption band (lmax ¼ 616 nm (shoulder), end-absorption @750 nm). Thus, the chromophoric properties of
204 6 Functional Conjugated Materials for Optonics and Electronics by Tetraethynylethene Molecular Scaffolding
Fig. 5. Extended TEE dimer, containing a long linear conjugation path (shown in bold) [29d].
TEE dimers are readily tunable upon aryl substitution, extending the absorption range into the near-IR region [29d].
The long linear conjugation in TEE dimers 20 and 24 causes almost a doubling of the second hyperpolarizabilities relative to that of TEE monomer 11 (Figure 3), containing the same number of aryl groups [29d]: g ¼ 2036 10 36 esu for 20, g ¼ 2243 10 36 esu for 24, measured by third-harmonic generation experiments at a wavelength of 1.907 mm (thirdharmonic wavelength: 636 nm). However, it is important to note that this increase in g corresponds to an increase in the molecular weight, leaving the macroscopic nonlinearities al-
most unaltered: wð3Þ100 %=wð3Þfs ¼ 519 for 20, wð3Þ100 %=wð3Þfs ¼ 570 for 24 (fs ¼ fused silica: wð3Þfs ¼ 1:16 10 14 esu).
6.4
Two-Dimensional Sca olding: Expanded Carbon Cores
Based on either cis-substituted or geminally substituted TEE derivatives, two families of macrocycles have been constructed, namely perethynylated dehydroannulenes and expanded radialenes.
6.4.1
Perethynylated Dehydroannulenes
Glaser–Hay macrocyclization of cis-bis(trialkylsilyl)-protected TEEs generated the perethynylated octadehydro[12]annulenes 26a,b and dodecahydro[18]annulenes 27a,b (Figure 6) with fully planar p-conjugated carbon cores as evidenced by X-ray crystallography [30]. The 4n þ 2 ðn ¼ 4Þ p electron cycles 27a,b possess a large HOMO-LUMO gap (2.57 eV in pentane) and are aromatic, as is the parent, unsubstituted dodedecahydro[18]annulene prepared by Okamura and Sondheimer in 1967 [31]. In contrast, the 4n ðn ¼ 3Þ p electron cycles 26a,b possess a small HOMO-LUMO gap (1.87 eV) and are anti-aromatic like the parent, unsubstituted octadehydro[12]annulene that was synthesized simultaneously by two different groups in 1966 [32, 33]. Electrochemical studies [29b, 30b] showed that [12]annulene
26b is more readily reduced than [18]annulene |
27b (E ¼ 0:99 and 1.46 V for 26b, |
E ¼ 1:12, 1.52, and 1.91 V for 27b, versus |
Fc/Fcþ, in THF). Indeed, 27b loses its |

6.4 Two-Dimensional Scaffolding: Expanded Carbon Cores 205
Fig. 6. Planar, perethynylated aromatic (27a,b) and anti-aromatic (26a,b) dehydroannulenes [30].
Hu¨ckel aromaticity upon reduction, whereas 26b gains aromaticity ð4n þ 2; n ¼ 3Þ when converted to its dianion. The e ect of substituting the peripheral silyl groups by aromatic groups still needs to be explored for these perethynylated dehydroannulenes.
6.4.2
Perethynylated Expanded Radialenes
Expanded radialenes are a family of macrocycles derived from radialenes [34] by the formal insertion of ethynediyl or buta-1,3-diynediyl moieties into the cyclic framework between each pair of vicinal exo methylene units. Geminally substituted TEEs, as well as TEE dimers, are perfect building blocks for the construction of perethynylated expanded radialenes. Thus, by bis-deprotection of a suitable bis-silylated TEE precursor, followed by oxidative coupling under high-dilution conditions, the expanded [n]radialenes 28a–c (n ¼ 3–5) were prepared (Figure 7) [29]. The expanded [6]radialene 28d, as well as the expanded [n]radialenes 29a,c ðn ¼ 4; 6Þ and 30a–c ðn ¼ 4; 6; 8Þ were prepared in a similar manner by cyclization of TEE dimers 21, 19, and 23, respectively, after their desilylation by treatment with tetrabutylammonium fluoride. The expanded [5]radialene 29b was obtained by cyclizing dimer 19, after bis-deprotection, with a related TEE trimer. A study of the electronic properties of these three di erently substituted series of expanded radialenes (R ¼ Si(iPr)3, 3,5-di(tBu)Ph, or p- (C12H25)2NPh) provided fundamental information regarding p-electron delocalization in cyclic systems, where both linear and cross-conjugative pathways can be operational [29d].
Fig. 7. Expanded radialenes bearing peripheral substituents of di erent electron-donating strengths [29].
206 6 Functional Conjugated Materials for Optonics and Electronics by Tetraethynylethene Molecular Scaffolding
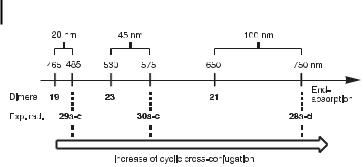
Fig. 8. Comparison of the end-absorptions of TEE dimers and expanded radialenes reveals that the degree of cyclic cross-conjugation in the radialenes increases with increasing donor strength of the peripheral substituents [29d].
The absorption spectra of the expanded radialenes were compared to those of their related TEE dimers, which serve as models for the longest linearly conjugated p-electron fragment in the macrocycles. Within each radialene series, the optical end-absorption remained almost constant, and hence was independent of the ring size. For the per(silylethynylated) expanded radialenes 29a–c, this end-absorption was seen to be bathochromically shifted by only @20 nm compared to that of dimer 19. However, the introduction of aryl substituents results in dramatic spectral changes (Figure 8). Thus, for the series 30a–c versus 23, a bathochromic shift of @45 nm is observed, and for the series 28a–d versus 21, the shift is as much as 100 nm. These large bathochromic shifts are interpreted as resulting from a high degree of macrocyclic cross-conjugation in the arylated macrocycles 28a–d and 30a–c. Thus, it transpires that arylation is of major importance for inducing macrocyclic crossconjugation, which becomes increasingly e cient with increasing donor strength of the substituents.
Another interesting property of expanded radialenes is their strong ability to accommodate electrons upon reduction, as revealed by an electrochemical study by Gross, Gisselbrecht, and Boudon [29d]. Thus, in all three series, the first reduction occurred at anodically shifted potentials relative to the reference TEE dimers; the following shifts were observed: þ170– 440 mV for 29a–c relative to 19, þ210–330 mV for 30a,b relative to 23, and þ240–320 mV for 28a–c relative to 21 (Figure 9). Thus, the radical anions of expanded radialenes are very stable, particularly those of the expanded [3]- and [4]radialenes, whatever the nature of the substituents.
Donor-substituted expanded radialenes also display huge nonlinear optical coe - cients: 28a: g ¼ 18000 10 36 esu, wð3Þ100 %=wð3Þfs ¼ 2700; 28b: g ¼ 7000 10 36 esu, fs ¼ 800 (wð3Þfs ¼ 1:16 10 14 esu). These values are, however, to some extent resonance-enhanced owing to absorption at the third-harmonic wavelength of 700 nm (laser
wavelength: 2.1 mm) [29c, 29d].
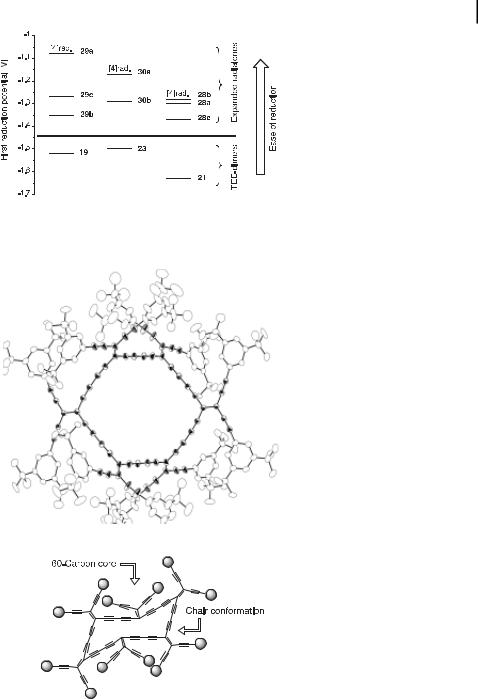
Interestingly, the perethynylated core of an expanded [6]radialene contains 60 carbon atoms and can therefore be viewed as an isomer of Buckminsterfullerene C60. X-ray crystal structure analysis of 30b (Figure 10) revealed that the cyclic core adopts a non-planar, ‘‘chairlike’’ conformation, with an average torsional angle of 57.2 [29d]. Each buta-1,3-diynediyl moiety only deviates slightly from linearity. Thus, the corresponding bond angles vary from 174 to 180 . The six individual TEE units are almost planar, with deviations of no more than 0.03 A˚ from the mean plane.
6.4 Two-Dimensional Scaffolding: Expanded Carbon Cores 207
Fig. 9. Comparison of first reduction potentials (vs. Fc/Fcþ) for the three di erently substituted series of TEE dimers/expanded radialenes [29d]. Solvent: THF for 19, 29a–c; CH2Cl2 for 21, 23, 28a–c, and 30a,b.
Fig. 10. X-ray crystal structure of expanded [6]radialene 30b; H-atoms have been omitted for clarity. Atomic displacement parameters at 203 K are drawn at the 20 % probability level. The perethynylated C60-core is highlighted in bold; it adopts a chair-like conformation [29d].

208 6 Functional Conjugated Materials for Optonics and Electronics by Tetraethynylethene Molecular Scaffolding
Fig. 11. ‘‘Superdiamond’’ network, an elusive allotrope of carbon.
The synthesis of all-carbon networks, representing artificial counterparts of the two naturally occurring allotropes, graphite and diamond, has attracted considerable interest in recent years [1c]. A fascinating allotrope, still elusive, is the ‘‘superdiamond’’ network depicted in Figure 11. It is tempting to compare this network with an expanded [6]radialene, recognizing that they contain chair-form cyclic parts of the same size, the only di erence being the hybridization of the corner carbon atoms, sp3 versus sp2. In this context, the class of perspirocyclopropanated macrocyclic oligo(diacetylene)s, [n]rotanes, should be mentioned. These macrocycles are structurally similar to the expanded radialenes, but have insulating sp3 carbon centers separating the buta-1,3-diynediyl fragments. Indeed, [6]rotanes also adopt chair-like conformations [35].
6.4.3
Cyclic Platinum s-Acetylide Complex of Tetraethynylethene
By Pt-TEE sca olding, the macrocyclic complex 31 (Figure 12) was prepared by deprotection and Hay coupling of dimer 32 [36]. This macrocycle can be envisaged as an expanded
Fig. 12. Linear and cyclic Pt-TEE complexes [36].
6.5 Linearly p-Conjugated Oligomers and Polymers: Poly(triacetylene)s 209
Fig. 13. Progression of linearly p-conjugated all-carbon backbones from trans-polyacetylene (PA) through trans-poly(diacetylene) (PDA), transpoly(triacetylene) (PTA), to carbyne.
[4]radialene containing two platinum centers as an integral part of the cyclic core. The end-absorption of 31 is bathochromically shifted relative to that of the dimer 32 and extends well beyond 500 nm to about the same onset as observed for 30a. Thus, it seems that there is electronic communication along the rectangular perimeter of 31.
6.5
Linearly p-Conjugated Oligomers and Polymers: Poly(triacetylene)s
Polyacetylene (PA) is the simplest conjugated polymer [37] with an all-carbon backbone not composed of aromatic rings (Figure 13), and has been extensively exploited for its electrical conductivity upon doping [38]. The poly(diacetylene)s (PDAs) and poly(triacetylene)s (PTAs) are the next representatives in a progression that ultimately leads to carbyne. PTAs were first synthesized in 1994 [39], after suitable monomeric tetraethynylethene and trans-1,2- diethynylethene (DEE) building blocks had become available [40]. The p-electron conjugation in the PTA backbone is best described as cylindrical and is maintained upon rotation about C(sp)aC(sp2) and C(sp)aC(sp) single bonds, since UV/vis studies on PTA oligomers 33a–e, 34a–c, and 35a,b, dendritically encapsulated by shells of Fre´chet-type dendrons [41], demonstrated that the electronic properties were not altered upon distorting the backbone out of planarity as a result of steric compression of the bulky dendritic wedges (Figure 14) [42].
Fig. 14. Dendritic PTA oligomers with the linearly p-conjugated allcarbon backbone encapsulated by dendrons of first to third generation [42].
210 6 Functional Conjugated Materials for Optonics and Electronics by Tetraethynylethene Molecular Scaffolding
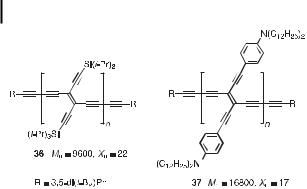
Fig. 15. PTAs containing lateral silyl [39] or anilino [29c] groups. Mn ¼ average molecular weight; Xn ¼ average number of TEE monomer units.
The question that needs to be addressed is how the physical properties of PTAs are influenced by aryl groups, in either the lateral positions or within the carbon backbone, or by donor–donor or acceptor–acceptor substitution in the end groups.
6.5.1
Lateral Aryl Substitution
The e ect of lateral aryl donor functionalization can be understood by comparing the two PTAs 36 [39] and 37 [29c], the first containing lateral silyl groups and the second lateral anilino groups (Figure 15). They were readily obtained by oxidative Glaser–Hay polymerization of trans-bis-deprotected TEEs in the presence of the end-capping reagent 3,5-di(tert- butyl)phenylacetylene. By 1H NMR signal integration, the average molecular weight Mn and the degree of polymerization Xn were determined. Comparison of their physical properties reveals a significant enhancement upon lateral donor functionalization. Thus, the solutionstate optical gap of 37 (Eg ¼ 1:6 eV) is substantially reduced relative to that of 36 (Eg ¼ 2:0 eV). The PTA 37 only showed one irreversible reduction at 1.28 V (versus Fc/Fcþ, in CH2Cl2), this being anodically shifted by þ350 mV relative to the first reduction of TEE dimer 21. Thus, the longer linear conjugation length in 37 facilitates the first reduction relative to that in 21. Moreover, as is generally observed for anilino-substituted TEE sca olds, 37 undergoes reversible oxidation centered on the anilino groups (E ¼ þ0:45 V versus Fc/ Fcþ in CH2Cl2).
6.5.2
Aromatic Spacer Units
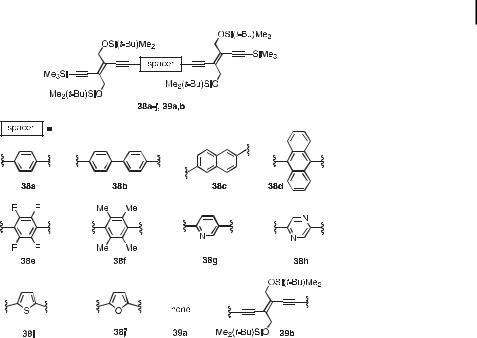
The modulation of the electronic properties of PTA oligomers by p-electron-deficient and -rich aromatic and heteroaromatic spacer units was first tested in DEE dimers 38a–j and 39a,b (Figure 16) [43]. The insertion of such spacers was found to have a profound influence on the molecular properties, such as the fluorescence behavior. Thus, whereas the quantum yields of compounds 39a,b and hybrid trimer 38j were below 5 % in chloroform (lexc ¼ 356 nm), hybrid derivatives 38a, 38e, 38g, and 38i exhibited quantum yields between 20 and
6.5 Linearly p-Conjugated Oligomers and Polymers: Poly(triacetylene)s 211
Fig. 16. Dimers of trans-1,2-diethynylethene (DEE) containing aromatic spacer units [43].
50 %. The quantum yields were even increased above 50 % in hybrid trimers 38b–d, 38f, and 38h.
Whereas DEE trimer 39b experiences a large bathochromic shift of the longest-wavelength absorption maximum relative to that of DEE dimer 39a (lmax ¼ 407 nm, shoulder (39b), lmax ¼ 376 nm (39a)), hybrid trimers 38a–c display about the same longest-wavelength absorption maxima, as well as end-absorptions, as 39a. Thus, benzenoid spacers are less e ective in transmitting p-electron delocalization along the oligomeric backbone. In contrast, introduction of the anthracene spacer in 38d produces a remarkable red-shift of all the bands in the UV/vis spectrum. Insertion of electron-rich heterocyclic spacers, as in thiophene derivative 38i and furan derivative 38j, results in very e cient p-electron delocalization, shifting the longest wavelength absorption maximum to 404 nm (shoulder) and 398 nm (shoulder), respectively. While a pyridine spacer (in 38g) does not produce a bathochromic shift, the pyrazine-spaced compound 38h exhibits a longest-wavelength absorption at lmax ¼ 392 nm (shoulder). Indeed, the benzenoid aromaticity is more pronounced in pyridine than in pyrazine. Pyridine derivative 38g represents an interesting example of a molecular system in which both the electron absorption and emission characteristics can be reversibly switched as a function of pH. Thus, protonation results in a large bathochromic shift of the most intense electronic absorption band from lmax ¼ 337 to 380 nm and a decrease in the fluorescence quantum yield from 40 % to 7 % (lexc ¼ 356 nm) [43c].
Based on the biphenyl spacer, a long-chain oligomer (40) was prepared (Figure 17) [43d]. This PTA retained the strong emission behavior of 38b, displaying a quantum yield of 44 % in chloroform. Its potential in light-emitting diodes is currently under investigation.
Recently, Pt-TEE molecular sca olding yielded PTA oligomers with [Pt(PEt3)2] as spacer units [44]. Both the linear and nonlinear optical properties of these compounds revealed an

212 6 Functional Conjugated Materials for Optonics and Electronics by Tetraethynylethene Molecular Scaffolding
Fig. 17. PTA comprised of biphenyl-spaced DEE units [43d].
almost complete lack of p-electron conjugation along the backbones, that is, the Pt groups act as insulating centers in these linear systems. This insulating behavior of the [Pt(PEt3)2] spacer units is in contrast to their ability to convey, at least to some extent, p-electron delocalization in the macrocyclic complex 31.
6.5.3
Donor–Donor and Acceptor–Acceptor End-Functionalization
The PTA mono-, di-, tri-, tetra-, penta-, and hexamers 41a–f and 42a–f (Figure 18) provide useful information regarding the influence of donor–donor (D–D) and acceptor–acceptor (A–A) substitution in the end groups of PTAs based on the trans-1,2-diethynylethene repeat unit [45]. As reference points for the infinitely long polymers, compounds 41g and 42g (@18 and 12 DEE units) were studied. From the longest-wavelength absorption maxima of the compounds in each of these two series, the e ective conjugation length (ECL) was estimated. The ECL indicates the number of repeat units required to furnish size-independent properties in a conjugated polymer. The electronic properties of the end caps were found to have a strong bearing on the ECL. Thus, the ECL of the A–A PTA oligomer 42g was determined to be n ¼ 10, this being of the same length as found for silyl group end-capped PTA oligomers. However, the D–D PTA oligomer 41g exhibits a much shorter ECL of only n ¼ 4. This decrease in ECL is probably explained by the appearance of strong CT bands that dominate the absorption properties. Raman scattering measurements suggested an even smaller value of n ¼ 3 for this PTA.
6.6
Conclusions
Tetraethynylethene molecular sca olding has provided conjugated materials with properties that are greatly enhanced upon incorporating functional arene units. Indeed, third-order nonlinear optical properties are increased by aryl groups; photochemical cis-trans isomer-
Fig. 18. PTAs containing either donor or acceptor end-caps [45].