

Astruc D. - Modern arene chemistry (2002)(en)
.pdf7.4 Solid-State Structures and Liquid-Crystalline Properties of the PPEs 233
170 C without attaining the isotropic phase. At room temperature, both polymers show a higher than nematic order, as is evident from their polarizing micrographs. The textures (Figure 2a) suggest an ordering of the lamellar smectic type. This ordering is unusual because the organometallic main chains cannot form a planar board-like structure. Investigation of the organometallic polymer 65a by transmission electron microscopy (TEM) revealed an ordered lamellar superstructure with a lamellar width of approximately 18 nm. The lamellae are several hundred nm long. The extended length of the polymer chain 65a is 18 nm. This result was based on the Pn value obtained by gel permeation chromatography, with the extended length of the macromolecule and the width of the lamellae conforming. The extended length of 65c is 21 nm, while the observed lamellae are 24 nm wide by TEM. According to powder and electron di raction data, the polymer chains must be aligned perpendicularly to the axis of the lamellae and are responsible for their width. These polymers show a highly ordered lamellar phase, although their ordering principle is fundamentally di erent to that observed for the dialkyl-PPEs (vide infra, Figure 4b).
7.4.2
Poly(dialkylparaphenyleneethynylene)s 12
High molecular weight PPEs obtained by ADIMET are stable and do not decompose up to 250 C in air and up to 300 C under nitrogen. The high stability is a testimony to both their
Fig. 4. Supramolecular arrangements of organometallic (65) and organic poly(aryleneethynylene)s (12) in the solid state.

234 7 The ADIMET Reaction: Synthesis and Properties of Poly(dialkylparaphenyleneethynylene)s
Fig. 5. Variable-temperature powder X-ray di raction of didodecyl-PPE 12c.
defect-free structure and the absence of electron-releasing oxygen substituents, which make the dialkoxy-PPEs so prone to oxidative decomposition at elevated temperatures. As a consequence of their high thermal stabilities, most ADIMET-made dialkyl-PPEs (didodecyl-, bis(ethylhexyl)-, and bis(3,7-dimethyloctyl)-substituted) 12c–f melt reversibly without decomposition. They attain the isotropic phase at temperatures in the range 100–250 C depending upon their Pn and the nature of their side chain. Increasing Pn and decreasing the length of the side chains raises the melting point [46, 47]. For example, dihexyl-PPE 12a with a Pn of 100 cannot be melted, but decomposes above 270 C with darkening.
The favorable stability of the ADIMET PPEs allowed a determination of their temperaturedependent X-ray di raction and optical microscopy data. Didodecyl-PPE 12c is crystalline at
Fig. 6. Single-crystal X-ray di raction analysis of 2,5-bis(ethylhexyl)-1,4-dipropynylbenzene 10d [48].

7.5 Spectroscopic Properties of Dialkyl-PPEs 12 235
Fig. 7. Packing model of didodecyl PPE 12c in the solid state. The packing was derived from single-molecule MM2 calculations utilizing powder di raction and electron di raction data obtained for the PPEs.
room temperature and shows di raction peaks in both the small-angle and wide-angle regions of the powder di raction spectrum (Figure 5). At 110 C, all of the wide angle peaks have disappeared and only the intense small-angle peak at d ¼ 25:0 A˚ remains. This suggests the transition into the smectic liquid-crystalline state. At 140 C, all of the di raction peaks have disappeared, an isotropic melt has formed, and samples of 12c appear black under crossed polarizers in optical microscopy. Di erential scanning calorimetry (DSC) confirms

236 7 The ADIMET Reaction: Synthesis and Properties of Poly(dialkylparaphenyleneethynylene)s
Fig. 8. Liquid-crystalline textures of didodecyl-PPE 12c. Bars represent 20 microns. These PPEs form homeotropic smectic phases; the textures are typical for an edge-on view of lamellae that show apparent nematic ordering in this projection [46].
the phase transitions. From X-ray/electron di raction data and single-crystal structures of a series of dipropynyldialkylbenzenes 10 (Figure 6), a valid packing model of PPEs (12) at ambient temperatures was developed. This is a refined version of the models suggested by Wrighton [43] and by Le Moigne [42]. Dialkyl-PPEs 12 form a doubly lamellar structure in the solid state [48]. The optimum space filling of the long alkyl chains forces the PPE backbone into planarity, and enables the packing of their backbones on top of one another. The polymer chains pack in an AB pattern, in which the positions of the vertically adjacent polymer chains are displaced by 3.5 A˚ to accommodate the spatial requirements of the side chain and to ensure optimum electrostatic interaction of the main chains as shown in Figure 7.
Above 110 C, this arrangement becomes mobile, and a smectic C liquid-crystalline phase is entered. Samples cooled down from the isotropic melt (140 C) show Schlieren and banded textures when viewed under crossed polarizers (Figure 8). These textures look similar to nematic Schlieren textures, but from the X-ray di raction data it is clear that 12c forms a homeotropically oriented smectic C phase. In a nematic phase, the small-angle di raction peak would be absent, and a broad scattering feature, a ‘‘nematic streak’’, would be observed. Polymer 12c was the first example of a PPE derivative for which three states of matter, i.e. crystalline, thermotropic liquid crystalline, and a highly viscous isotropic liquid, were accessible [46].
7.5
Spectroscopic Properties of Dialkyl-PPEs 12 [47–49]
An important point in the science of conjugated polymers is the dependence of their optical properties as a response to their physical state (solution, aggregated, liquid crystalline, solid state, thermal treatment) [50]. Processable PPEs, particularly dialkoxy-PPEs, have been known since the early 1990s [1], and their spectroscopic behavior has been studied both in

7.5 Spectroscopic Properties of Dialkyl-PPEs 12 237
good solvents (chloroform) and in the solid state. Early on, it was noticed by Wrighton, Weder, and Swager [43] that the absorptions of PPEs in the solid state were red-shifted from those observed in solution. Annealing of thin dialkoxy-PPE films at elevated temperatures led to a further red shift of the primary absorption band [43]. The underlying reason for this phenomenon was not well explained nor examined in depth [1]. The dialkoxy-PPEs are not well-behaved with respect to aggregation-induced changes in their optical properties and might not be the best examples for studying aggregation behavior by electronic spectroscopy. However, Swager has recently re-examined these issues utilizing self-assembled monolayers at the air–water interface (Langmuir–Blodgett technique) in order to gain a better understanding of the electronic properties of dialkoxy-PPEs [51].
There has also been discussion about the origin of the spectroscopic signature of dialkylPPEs upon aggregation, both in absorption and emission. It is clear for PPEs that excimer and aggregate formation have a substantial influence on their emission properties in the solid state. However, the contribution of multichain phenomena to the UV/vis spectra of the aggregated phases of dialkyl-PPEs is not clear. For the dialkyl-PPEs 12, a strong case can be made for conformational changes of single polymer chains inducing the ‘‘aggregation band’’ observed at 439–445 nm (Figure 9).
7.5.1
UV/vis Spectroscopy of Dialkyl-PPEs 12
The UV/vis spectra of 12 are broad and featureless in chloroform, showing an absorption maximum at 388 nm. If methanol is added, the colorless and purplish-emitting solutions turn yellow and almost non-emissive, but remain clear (Figure 9) [48, 49]. Similarly, thin films of 12e (Figure 10) form highly viscous melts above 190 C, which are almost colorless and their UV/vis spectra resemble those obtained for PPEs in chloroform solution at ambient temperatures.
Addition of methanol to a solution of 12c gives rise to a new band at 442 nm and a resolution of the high energy features into three bands at 416, 407, and 382 nm (Figure 9). The spacing between the 442 nm band and the other bands is 3554, 1928, and 1414 cm 1, which is not readily interpretable in terms of defined molecular vibrations. The energetic separa-
Fig. 9. Solvatochromic behavior of didodecyl-PPE 12c upon addition of methanol.
238 7 The ADIMET Reaction: Synthesis and Properties of Poly(dialkylparaphenyleneethynylene)s
Fig. 10. Thermochromicity of a co-hexyl-dodecyl-PPE 12e.
tion of the high energy features translates into vibronic spacings of 2140 cm 1 (382 ! 416 nm) and 1608 cm 1 (407 ! 382 nm), respectively, which can be interpreted as vibronic fine structure resulting from the CaC triple bond stretching (2140 cm 1) and an aromatic stretching (1608 cm 1) vibration.
What is the underlying reason for the the sharp and red-shifted band observed for dialkylPPEs 12 at 439–442 nm upon aggregation and in the solid state? This band could have several di erent origins: it could be (i) of strictly intramolecular nature, due to conformational processes of single polymer chains, (ii) of intermolecular nature, i.e. true aggregate formation between two or more polymer chains in a process that is similar to the formation of J-aggregates in the cyanines, (iii) a mixture of interand intramolecular processes, or (iv) a property of a single chain that is nevertheless elicited by aggregation and involves several chains resulting in a conformational change of single chains. The 439 nm band would then be an aggregate-induced band.
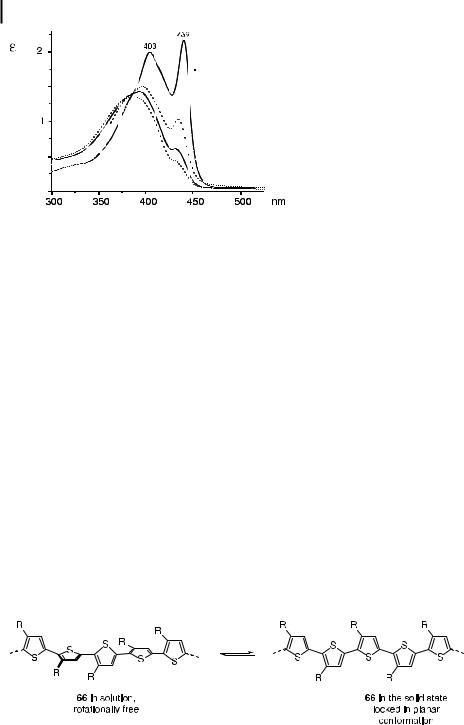
To explain the optical properties of the PPEs, comparison of their behavior with that of other conjugated polymers was helpful. The poly(3-alkylthiophene)s 66 show a similar spectral behavior (but in a more bathochromic regimen) as the PPEs. Thus, on entering the aggregated phase or on forming thin films, a substantial red shift of lmax and a significant sharpening of the bands is seen for 66. Wudl and Heeger [52] explained the spectroscopic changes in the poly(3-alkylthiophene)s 66 in terms of single chain planarization.
To confirm the qualitative agreement between the spectra of the polythiophenes and those of the PPEs, temperature-dependent UV/vis spectra of 12e were measured (Figure 10). The UV/vis spectrum recorded at ambient temperature shows the typical solid-state feature at
Fig. 11. Conformational preference of the poly(alkylthiophene)s according to Wudl et al. [51].

7.5 Spectroscopic Properties of Dialkyl-PPEs 12 239
439 nm. When the film of 12e was heated, the band at 439 nm slowly diminished and completely disappeared at temperatures above 185 C, at which point the spectrum resembled those obtained in dilute chloroform solution. A variable-temperature powder di raction study showed that 12e loses all its ability to di ract or scatter X-rays at temperatures above 190 C. Under crossed polarizers, films of 12e appear ‘‘black’’ above 190 C, indicating that this PPE transits from the liquid-crystalline form to the isotropic melt at this temperature.
To understand the implication of this experiment, knowledge of the conformational preference of diphenylacetylene in solution and in the solid state is necessary. According to Wudl, ‘‘we chemists are married to the concept of rotational invariance of modules connected by alkynes’’ [53]. This holds true in solutions of phenyleneethynylene oligomers and polymers, for which the barrier to rotation about the triple bonds is small (a1 kcal mol 1) according to quantum chemical calculations [54] and photoacoustic measurements [55]. However, this picture is no longer correct for solid PEs, where full planarization occurs even in the case of diphenylacetylene [56]. While the di erence between the planar and twisted conformations is too miniscule to prohibit deplanarization of PE oligomers in solution, the small energy di erence is su cient in the solid state to force them to adopt structures in which they have planar backbones. This e ect is more pronounced in the case of alkylsubstituted PEs, where the accommodation of the side chains reinforces and ‘‘freezes’’ the coplanar packing of the conjugated backbones.
While deplanarization has a small e ect on the total energy of the phenyleneethynylenes, its influence on the frontier orbitals (HOMO and LUMO), which are mostly responsible for the optical properties of conjugated molecules, is much more dramatic. Semiempirical calculations on an octameric phenyleneethynylene model compound, utilizing the AM1 and PM3 methods, revealed a significant increase in the band gap on going from the fully planar form to the more twisted forms (Figure 12). The band gap was calculated to be largest when all adjacent benzene units were twisted by 90 . In collaboration with Garcia-Garibay [57], a phenyleneethynylene model trimer was carefully analyzed, revealing a small band gap in the solid state and a larger gap in solution. A semiempirical calculation including configuration interaction supported the experimental evidence that planarization leads to an increased lmax in the UV/vis spectrum of phenyleneethynylenes.
A self-consistent picture arises, in which the optical changes of the PPEs upon thin-film formation can be attributed to an aggregation-induced planarization of single polymer chains. While the aggregation process involves many chains that enforce their mutual pla-
Fig. 12. Conformational preferences of dialkyl-PPEs 12 in solution and in the solid state.

240 7 The ADIMET Reaction: Synthesis and Properties of Poly(dialkylparaphenyleneethynylene)s
Fig. 13. (a) Overlay of absorption and emission spectra of 67 in chloroform at room temperature. (b) Overlay of absorption and emission spectra of 67 in low molecular weight polystyrene at 77 K.
narization invoking Ockham’s razor, the spectroscopic changes of dialkyl-PPEs 12 can be explained in terms of conformational changes of single polymer chains (Figure 12). However, interchain interactions may also play a minor role. PPEs bearing donor and/or acceptor substituents are fundamentally di erent because charge-transfer e ects will enhance their interchain interactions, and the influence of these on their optical properties will be more significant [50].
7.5.2
Fluorescence Spectroscopy : The Excited State Story [58]
The UV/vis spectra of 67 and 12a–f are sharp in the solid state, but broad and featureless in good solvents. This is due to the fact that the rotation of the PE units about the CcCaAr single bonds is unrestricted, and deviation of planarity between PE units in single chains increases the HOMO-LUMO gap [47]. The emission spectra of 12 and 67 are narrow and show a well-resolved shoulder (Figure 13a). For rigid molecules such as 67, one would expect approximate mirror symmetry of absorption and fluorescence with a relatively small Stokes’ shift. Thus, the question arises as to why this is not the case.
The optical and emissive properties of 12 and 67 are almost identical, confirming that 67 has reached the convergence length of PPEs and is thus a good model for 12. The use of 67 avoids issues of energy migration along single polymer chains, chain entanglement, energy transfer between multiple chains, and excimer formation. The optical behavior of PPEs may therefore be explained by investigating 67. When a solution of 67 in polystyrene is cooled to 77 K, its absorption spectrum narrows slightly. However, significant changes are seen in the emission spectrum (Figure 13b), which, according to Berg et al. [58], is broadened towards the blue side. In a time-resolved fluorescence experiment (Figure 15; 297 K, chloroform), the blue component of the emission decreases over time and the narrow fluorescence spectrum is reconstituted.
These experiments are consistently interpretable. The ground state of 67 features a shallow potential energy profile with respect to rotation about the PE units (CHCl3 solution at 297 K; Figures 13 and 14). In the excited state, this may not be the case. The spectroscopic data can be explained if the potential energy profile of 67* (Scheme 20) is much steeper than that of 67, and symmetric with respect to rotation (Figure 13a). This makes sense: in a va-

7.5 Spectroscopic Properties of Dialkyl-PPEs 12 241
Scheme 20
lence bond picture, the excited state of 67 has a diradicaloid/cumulenoid character and rotation about the PE units is not facile anymore because the allenic structure has a strong preference for planarity of the conjugated backbone, eliminating the twisted double bonds.
In good solvents at ambient temperature, the excited state (67*) will quickly relax to the planar form, so that only the 0–0 emission from S1 is detected in steady-state emission. If the same experiment is performed at low temperature and in a viscous solvent, the molecular torsion of 67* in attaining its planar form is hampered by the medium, and planarization is slow on the timescale of the fluorescence lifetime (355 ps). Emission will not only occur from the potential minimum of the lowest excited state, but from virtually all frozen rotamers resulting in a broad and blue-shifted spectrum. Only after planarization of 67* is complete narrow emission from the lowest excited-state conformation will reoccur. Consequently, planarization of the excited state rather than energy migration is likely to govern the emission behavior in PPEs such as 12.
In linear coupling models, the lowest-energy configuration is di erent in the ground and excited states, but the restoring forces are identical. The strong torsional coupling between the ground and excited states of 67 features a restoring force that is much stronger in the excited state than in the ground state, although the lowest-energy configuration is very similar. This situation is called quadratic coupling [58]. Quadratic coupling mimics spectroscopic e ects in PPEs that have been attributed to energy migration, mode mixing, and other processes. It may be important but unrecognized in other conjugated polymers [52].
Fig. 14. (a) Potential energy profile of 67 in the excited state with respect to the torsional angle about the spasp2 single bond. Note that the excited state has a much steeper torsional profile than the ground state. (b) Expected time-dependence of the emission in a system for which quadratic coupling is significant.

242 7 The ADIMET Reaction: Synthesis and Properties of Poly(dialkylparaphenyleneethynylene)s
Fig. 15. Time-resolved spectrum of the emission of 67 in chloroform at 298 K.
7.6
Self-Assembly of PPEs on Surfaces: From Jammed Gel Phases to Nanocables and Nanowires
PPEs 12 are rigid and linear conjugated organic polymers, and it is expected that due to their shape anisotropy, self-assembly processes of PPEs will be facile. In bulk PPE samples, solidstate organization leads to lamellar phases with high order, whereas in thin films additional supramolecular features may be possible. Casting a dilute yellow solution of PPE in toluene onto a silicon wafer followed by slow evaporation of the aromatic solvent led to thin films that were investigated by atomic force microscopy (AFM). AFM showed two di erent selfassembly modes in these thin films. One is on the 1–2 nm, i.e. molecular scale and corresponds to the lamellae observed by powder di raction analysis. The second features are much larger in their dimensions (Figure 16a). Long, cable-like structures form, that are approximately 30 nm in diameter and between 700 nm and 2 mm in length. Samori, Mu¨llen, and Rabe [59] had earlier observed that PPEs 12a display a rich self-assembly, and these authors demonstrated that depending on the conditions under which these films were grown
Fig. 16. (a) Nanocable arrangement of didodecyl-PPE 12c on a silicon wafer [60]. The observed superstructure exceeds the size of single lamellae. (b) Nanowires and (c) nanoribbons of PPEs 12a as observed by Samori, Mu€llen, and Rabe [59]. Single polymer chains are visible in
(c).