

Multiple Bonds Between Metal Atoms / 09-Ruthenium Compounds
.pdfRuthenium Compounds 407
Angaridis
Mononuclear cleavage products of the type Ru(O2CR)2(PPh3)2 and Ru(O2CR)2(CNBut)4 are obtained from reactions with Ph3P and ButNC, respectively. A variety of mononuclear Ru carbonyl compounds are also isolated from reactions with CO. Surprisingly, excess pyridine also reacts with the Ru2(O2CR)4 (R = Me, CF3) compounds to give the cleavage products Ru(O2CR)2(py)4. With Ru2(O2CCF3)4, but not the other Ru2(O2CR)4 compounds, MeCN also causes cleavage to give [Ru(O2CCF3)2(MeCN)5]O2CCF3.
Reactions of Ru2(O2CR)4 compounds with bifunctional N-donor ligands, such as pyz, phz, and DMDCNQI, result in the formation of one-dimensional polymeric chain struc- tures.156-158 More extended architectures are obtained when polyfunctional ligands are used, as shown in the reaction of Ru2(O2CCF3)4 with TCNQ which gives a two-dimensional network [Ru2(O2CCF3)4]2(µ4-TCNQ).159 However, upon reaction of Ru2(O2C(CH2)6CH3)4 with the polyfunctional ligand TCNE a redox reaction occurs instead.156
Electrochemical measurements show that Ru24+ tetracarboxylates are easily oxidized to their Ru25+ analogs: a reversible or a quasi-reversible one electron oxidation is observed close to a potential where reduction of the Ru25+ species takes place. A study of a series of Ru2(O2CR)4 (R = H, Me, CH2Cl, Et, Ph, CF3) compounds reveals that the oxidation potentials are highly dependent on the solvent and the substituent R of the carboxylate bridges.80,144 For solvents that can axially coordinate to the Ru24+ unit, the stronger the coordination of the donor solvent, the easier the oxidation becomes. Furthermore, substituents R with strong electron withdrawing ability result in more difficult oxidations. For example, the oxidation of Ru2(O2CCH2Cl)4 in THF is more difficult than the oxidation of Ru2(O2CEt)4 with potentials of +0.29 V and -0.03 V, respectively, whereas Ru2(O2CCF3)4 shows a much more positive oxidation potential in acetone at +1.03 V.
The electronic structure of compounds of this type has been quite controversial. Room temperature magnetic susceptibility measurements on Ru2(O2CR)4 and Ru2(O2CR)4L2 complexes showed magnetic moments in the range of 1.9-2.2 BM, which indicates the presence of two unpaired electrons.144 This means that the electronic configuration can either be μ2/4β2/*3β*1 or μ2/4β2β*2/*2. Early theoretical calculations at the SCF-X_-SW level performed for Ru2(O2CH)4 predicted the former,60 while ab initio Hartree-Fock calculations conducted later led to the conclusion that the ground state can be better described by the latter.154 In each case the difference in the calculated energies of the two electronic configurations is too small to allow a definite assignment of the ground state. In addition, regardless of their paramagnetic nature, no EPR spectra have been observed for this type of compounds (probably due to large zero-field splitting),80,144 while PES studies on Ru2(O2CH)4 and Ru2(O2CCF3)4 have been inconclusive.153-155
Structural data support the μ2/4β2β*2/*2 electronic configuration. Considering that the Ru–Ru distances of Ru24+ tetracarboxylates are similar to those of the corresponding Ru25+ compounds, for which the electronic configuration is μ2/4β2(/*β*)3, it is expected the additional electron in the Ru24+ tetracaboxylates should enter a β* molecular orbital. The lengthening of the Ru–Ru bond caused by an additional electron in a β* molecular orbital is very small and can be counterbalanced by the decrease of the electrostatic repulsion between the Ru centers (lower mean oxidation state). It should be noted that addition of an electron to a /* molecular orbital should result in a substantial lengthening of the Ru–Ru bond. Recent DFT calculations predict a ground state electronic configuration in agreement with that suggested by the structural data.160
Variable temperature magnetic susceptibility studies have also been helpful in the assignment of the ground state electronic configuration. A study on Ru24+ long-chain alkyl tetracarboxylates over the temperature range of 6-400 K concluded that the ground state has a singlet component and that there is a thermally accessible triplet excited state, but it was
408Multiple Bonds Between Metal Atoms Chapter 9
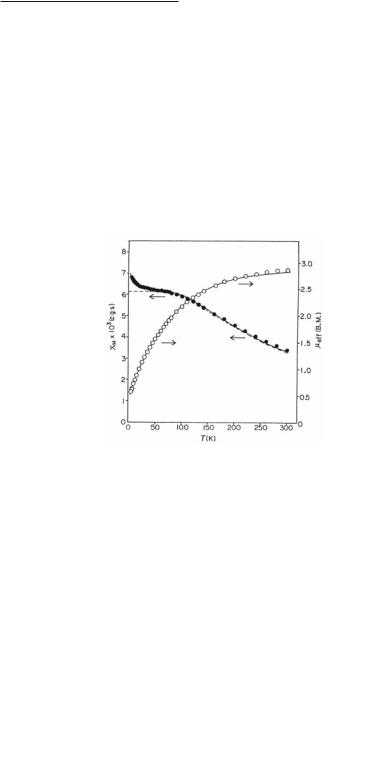
not possible to distinguish between the two possible ground state electronic configurations mentioned above.150 However, another study provided adequate and very persuasive evidence for the μ2/4β2/*2β*2 electronic configuration.148 The temperature dependence of the magnetic susceptibility of the complexes Ru2(O2CMe)4 and Ru2(O2CPh)4 over the temperature range of 6-298 K showed that the room temperature magnetic moment of ~2.8 BM per Ru2 unit tends towards zero as the temperature is lowered (Fig. 9.23). This implies a non-magnetic ground state at low temperatures, despite the fact that there are unpaired electrons at room temperature. This behavior is consistent with a /*2β*2 electronic configuration that results in a 3A2g ground state, which in turn splits under spin-orbit coupling into an 3Eg state with mS = ±1 and a much lower in energy A1g state (mS = 0) (9.5). The two states are separated by a large zerofield splitting (a value of ~250 cm-1 was calculated for the zero-field splitting parameter, D). As shown in Fig. 9.23, there is an excellent agreement of this model and the experimental data.
Fig. 9.23. Plots of the molar magnetic susceptibility and effective magnetic moment versus temperature for Ru2(O2CMe)4.
MagneticmeasurementsconductedforthepolymericcompoundsRu2(O2C(CH2)10CH3)4(pz)161 and Ru2(O2CCF3)4(phz)157 show that in both compounds there is an appreciable contribution of a large zero-field splitting arising from the S = 1 ground state to the resulting magnetic moments (D = 250-300 cm-1). However, for the former the data were inconclusive as to whether any interdimer antiferromagnetic coupling exists, while the data for the latter suggest that the Ru24+ units are weakly antiferromagnetically coupled with a coupling constant of -3 cm-1. Other Ru24+ tetracarboxylates linked by pyz, 4,4'-bipy, and dabco have also been studied, and show no interdimer interactions.162
9.5

Ruthenium Compounds 409
Angaridis
The polymeric compound with a two-dimensional network structure [Ru2(O2CCF3)4]2(µ4- TCNQ) exhibits a low magnetic moment at room temperature (ρMT = 0.678 cm3Kmol-1) which decreases as the temperature approaches 0 K.159 Although the contribution of the zerofield splitting to the decrease of the magnetic moment at lower temperatures is significant, the data are in accordance with the existence of a strong antiferromagnetic interaction between the Ru24+ units.
The complexes with axially coordinating nitroxide radicals Ru2(O2CCF3)4(tempo)2 and Ru2(O2CC6F5)4(tempo)2, which have a large zero-field splitting within the dimetal unit (D ~240 cm-1), display strong antiferromagnetic interactions between the Ru2 core and the nitroxide radical with J2 = -263 and -234 cm-1, respectively,163 which are much larger than those in the Ru25+ analog [Ru2(O2CCMe3)4(tempo)2]+.73 No coupling was observed between the two axially coordinating tempo ligands (J1 = 0).
9.3.2 Ru24+ compounds with N,O-donor bridging ligands
Oxopyridinate ligands
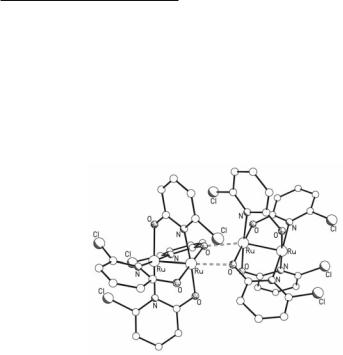
Prior to the isolation of any other Ru24+ compound, Ru2(mhp)4 (Fig. 9.24) was prepared in low yield (8%) from the reaction of Ru2(O2CMe)4Cl with Na(mhp) in 1981.164 It was later shown that by employing a Ru24+ tetracarboxylate instead of a Ru25+ tetracarboxylate as starting material in such a reaction, higher yields of Ru2(mhp)4 can be obtained.165 Other Ru24+ tetraoxopyridinates, like Ru2(chp)4, Ru2(fhp)4, and Ru2(bhp)4, have been synthesized following a similar synthetic strategy, i.e., from the reactions of Ru2(O2CMe)4 with either an excess of the molten hydroxypyridines, or stoichiometric amounts of their Na+ salts (methods which are comparable to those employed for the synthesis of analogous Cr24+ and Mo24+ compounds).
The majority of Ru24+ tetraoxopyridinates exist as discrete paddlewheel complexes, which do not associate through the axial positions to form polymers, except for some cases in which dimerization occurs (see below). Axial ligands are usually coordinating solvent molecules. The Ru–Ru bond lengths span from 2.235 to 2.274 Å (Table 9.2), and they do not show any dependence on the bridging oxopyridinate ligand and the steric effect of the coordination mode. However, axial ligation results in longer Ru–Ru bonds.
Fig. 9.24. The structure of Ru2(mhp)4.
The major factor determining the preferred regioisomer for the Ru44+ tetraoxopyridinates is not the axial ligation as in Ru2(Xhp)4Cl compounds, but the size of the X group in the Xhp ligand. For large X groups, like Br and Me, the trans-(2,2) regioisomers form, since only two
410Multiple Bonds Between Metal Atoms Chapter 9
large substituents X can be accommodated at each end. In contrast, for smaller substituents, like F and Cl, the polar arrangements (4,0) and (3,1) are preferred. In this latter case any steric hindrance that might be imposed due to the X substituents at one axial site can be overcome by the stabilization gained from the coordination of a ligand to the unencumbered axial site. For example, in the (4,0) regioisomer of Ru2(fhp)4 a THF molecule coordinates to the open axial position.166 Stabilization can also be gained through dimerization. For example, Ru2(chp)4, when isolated as the (3,1) regioisomer, dimerizes in the absence of coordinating solvents as shown in Fig. 9.25.165 The last compound has also been isolated as the trans-(2,2) regioisomer.98
Fig. 9.25. The structure of the (3,1) regioisomer of Ru2(chp)4.
The Ru2(Xhp)4 complexes exhibit room temperature magnetic moments of ~2.5 BM, indicative of two unpaired electrons.165 As for the Ru24+ tetracarboxylates, there are two possible electronic configurations, μ2/4β2/*3β*1 or μ2/4β2β*2/*2. The PES of Ru2(mhp)4 shows three peaks of approximately equal intensities at ionization energies of 5.8, 6.3, and 6.8 eV.164 These energies were assigned to /*, β*, and β, respectively. It was asserted that this spectrum “suggests” a β*/*3 electronic configuration. However, it is equally compatible, if not more so with a β*2/*2 electronic configuration (because the β and β* peaks are of about equal intensity rather than in a 2:1 ratio).
Structural data clearly favor the μ2/4β2β*2/*2 electronic configuration since the Ru–Ru distances in Ru44+ oxopyridinates (2.235 to 2.274 Å) fall in the range of the Ru2(O2CR)4Cl compounds, which are known to have two /* electrons. Additional support for the electronic configuration is provided by variable temperature magnetic measurements. The complexes Ru2(mhp)4, Ru2(chp)4, Ru2(bhp)4, and Ru2(fhp)4 exhibit similar magnetic behavior165,166 with room temperature magnetic moments of ~2.5 BM that drop to an extrapolated value of 0 BM as the temperature approaches 0 K, as in the Ru2(O2CR)4 compounds. This behavior is not consistent with a /*3β*1 configuration or a singlet-triplet Boltzmann distribution based on /*3β*1 and /*4 electronic configurations, since these would lead to qualitatively different types of behavior as a function of temperature. However, the magnetic data are consistent with a ground state derived from μ2/4β2β*2/*2 configuration, which results in a 3A2g state that is split by spin-orbit coupling (D ~ 200-250 cm-1) to give a lower state with Ms = 0.
Quantitative support for the above mentioned electronic configuration comes from SCF-X_ theoretical calculations for the Ru2(Xhp)4 compounds, in which the Xhp ligand was modeled by the ONHCH fragment.166

Ruthenium Compounds 411
Angaridis
9.3.3 Ru24+ compounds with N,N'-donor bridging ligands
Formamidinate ligands
The Ru24+ tetraformamidinates are usually synthesized by ligand metathesis reactions of Ru2(O2CMe)4 with stoichiometric amounts of Li+ salts of formamidinates.167 Alternatively, they can be synthesized from their Ru25+ analogs either by bulk electrolysis,168 or by reduction with Zn.129 They are isolated as air-sensitive solids which give normal 1H NMR spectra.
Upon reactions with Lewis bases which are also strong /-acceptors, like CO, Ru2(DArF)4 compounds give axial adducts without disruption of the Ru–Ru bond, in contrast to their carboxylate analogs which react with CO to decompose to mononuclear species. For example, Ru2(DPhF)4 reacts with CO to give Ru2(DPhF)4(CO).168 Attempts made to isolate the bis-CO adduct have been unsuccessful.
The cyclic voltammogram of Ru2(DTolF)4 shows two redox processes: a reversible oxidation at +1.163 V and a reversible reduction at -0.118 V. However, the electrochemical behavior of this compound is not very well understood, since the oxidation potential suggests that it should be stable towards oxygen, which is not true.167
In the solid state Ru2(DArF)4 compounds exist as discrete molecules which do not associate, as shown by the structure of Ru2(DTolF)4 (Fig. 9.26).167 There are only three crystallographically characterized complexes of this type (Table 9.2). Two of them, Ru2(DAniF)4 and Ru2(DTolF)4, exhibit similar Ru–Ru bond lengths at 2.454(1) and 2.474(1) Å, respectively. However, the third one, Ru2(DPhF)4(CO), displays a much longer Ru–Ru bond length of 2.554(1) Å, which is comparable with the distances observed in Ru2(O2CEt)4(NO)2 and Ru2(O2CCF3)4(NO)2.80
The Ru–Ru distances of Ru24+ tetraformamidinates are the longest observed among the Ru24+ paddlewheel complexes. Even though it has been proposed that this might be due to the larger “bite” angle of the bridging formamidinates, the real reason is electronic in nature. Generally, for Ru24+ compounds there are three possible electronic configurations: μ2/4β2/*4, μ2/4β2/*3β*1, μ2/4β2/*2β*2, depending on the ordering of the /* and β* molecular orbitals levels and their energy separation. The diamagnetism of Ru2(DArF)4 compounds (as indicated by their normal 1H NMR spectra) together with the long Ru–Ru distances suggest that the frontier electrons are paired in the strongly antibonding /*, rather than in the weakly antibonding β* molecular orbital, as the lengthening of the Ru–Ru bond caused by the pairing of electrons in the β* molecular orbital would have been very small. As a result, the μ2/4β2/*4 electronic configuration was proposed.
Fig. 9.26. The structure of Ru2(DTolF)4.

412Multiple Bonds Between Metal Atoms Chapter 9
SCF-X_ theoretical calculations on the Ru2(HNCHNH)4 model compound support the above mentioned electronic configuration.167 The formamidinate ligands interact with the Ru24+ core raising the energy of the β* molecular orbital above that of the /* orbital. As a result, the HOMO is a fully occupied /* molecular orbital and the LUMO is a β* molecular orbital. The /*–β* separation, calculated at ~1.18 eV, is large enough to prevent any appreciable population of higher magnetic states at room temperature.
Triazenate ligands
The Ru24+ tetratriazenates can be synthesized from the stoichiometric ligand metathesis reactions of Ru2(O2CMe)4 with Li+ salts of triazenates (DArTA).80 They are isolated as air-stable solids which give normal 1H NMR spectra.
The Ru2(DArTA)4 complexes generally do not react with weak Lewis bases (e.g., THF, acetone, MeCN) to give axial adducts; however, Ru2(DTolTA)4 gives a mono-MeCN adduct.169 Similarly to their formamidinate analogs, they react with Lewis bases which are also strong /-acceptors to form adducts. For example, Ru2(DPhTA)4 reacts with NO and CO to form strong bis-adducts and with the bulkier ButNC to form a mono adduct. However, it does not react with py nor PPh3. This lack of reactivity is almost certainly due to steric constraints imposed by the bulky phenyl groups of the DPhTA ligands.80
Cyclic voltammetry measurements of Ru2(DPhTA)4 show three redox processes. The NO, CO, and ButNC axial adducts of Ru2(DPhTA)4 show similar redox behavior.80 For the latter complexes the potentials of the reduction and the first oxidation processes vary considerably, which gives an indication that these are metal-based processes corresponding to Ru24+ + e- Α Ru23+ and Ru24+ Α Ru25+ + e-, respectively. However, the second oxidation wave appears almost invariantly at the same potential (~ +1.30 V), which suggests that this redox process may be associated with the ligand and not with the dimetal core.
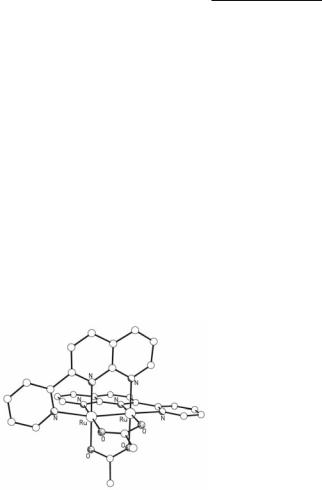
In the solid state, Ru24+ tetratriazenates exist as discrete molecules which do not associate, as shown by the structure of Ru2(DTolTA)4 in Fig. 9.27.170 The Ru–Ru bond lengths lie in the range of 2.399 to 2.417 Å (Table 9.2). Although shorter than those in the Ru24+ tetraformamidinates, these distances are significantly longer than those of most of the Ru24+ paddlewheel compounds. Interestingly, the Ru–Ru bond length of 2.407(1) Å in Ru2(DTolTA)4(MeCN)169 is slightly shorter than the corresponding distance of 2.417(2) Å in Ru2(DTolTA)4.170 For most Ru2 compounds, axial ligation causes an elongation of the M–M bond distance, since the μ donation of the ligand increases the anti-bonding μ* electron density between the two metals. In this case it appears that along with the μ donation of the axially coordinated MeCN, there is a moderate /-back donation from the /* metal orbitals to the empty /* orbitals of MeCN, which partially cancels the lengthening of the Ru–Ru bond distance caused by μ donation.
Fig. 9.27. The structure of Ru2(DTolTA)4.
Ruthenium Compounds 413
Angaridis
The long Ru–Ru bond lengths of Ru24+ tetratriazenates together with their diamagnetism (as indicated by their normal 1H NMR spectra) suggest the μ2/4β2/*4 electronic configuration. This is supported by SCF-X_ theoretical calculations carried out on the simplified computational model Ru2(HNNNH)4, which show a strong interaction between the β* orbital of the Ru24+ core and the p/ lone pair of the ligands.171 The β* molecular orbital is higher in energy than the /* molecular orbital by ~1 eV. The large /*–β* separation indicates that the β* is thermally inaccessible at room temperature, resulting in a singlet ground state.
Naphthyridine ligands
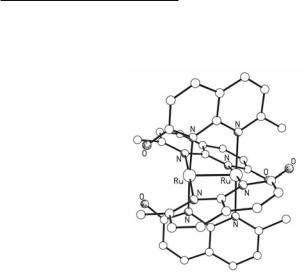
The Ru24+ naphthyridine compounds are synthesized by reacting Ru2(O2CMe)4Cl and excess of naphthyridines (or their Na+ salts) either in molten naphthyridines, or by prolonged reflux in MeOH, a process that causes the reduction to a Ru24+ core. For the neutral naphthyridines to replace negatively charged acetate groups, suitable counter ions (e.g., PF6-) are required.172 When naphthyridines with substituents that can coordinate axially to the dimetal unit are used, only partial substitution of the acetate groups of Ru2(O2CMe)4Cl takes place. For example, in the complexes cis-[Ru2(O2CMe)2(pynp)2](PF6)2172 (Fig. 9.28) and [Ru2(O2CMe)3(bpnp)]PF6 173 the substituents at the 2 and 7 positions of the bridging naphthyridine ligands block the axial positions preventing further substitution.
Fig. 9.28. The structure of the cation in [Ru2(O2CMe)2(pynp)2](PF6)2.
In the case of the naphthyridinone ligand mephonp, which can adopt either the N,O or the N,N' coordination mode, while in the Ru25+ complex trans-Ru2(O2CMe)2(mephonp)2Cl the mephonp ligands prefer the N,O coordination mode, in the Ru24+ analog trans- Ru2(O2CMe)2(mephonp)2 the N,N' coordination mode is adopted.137 The same preference for the N,N' coordination is observed in Ru2(meonp)4, although there is a twist of ~18º from the eclipsed configuration due to the steric requirement of the methyl substituents of the meonp ligands (Fig. 9.29).174 However, in the analogous complex Ru2(mephonp)4 the crowding of the adjacent phenyl substituents of the mephonp ligands allows only three of the bridging naphthyridinone ligands to adopt the N,N' coordination mode, while the fourth one is N,O-coordinated.137
Electrochemical data for Ru24+ naphthyridines show multiple redox processes due to both the Ru24+ core and the naphthyridine ligands. For example, the cyclic voltammogram of cis- [Ru2(O2CMe)2(pynp)2](PF6)2 shows four reversible, one-electron, ligand-based reductions and an irreversible, one-electron, metal-based oxidation at ~ +0.85 V.175 Free pynp exhibits a single two-electron reduction. However, in cis-[Ru2(O2CMe)2(pynp)2](PF6)2 the two-electron process for each one of the two ligands is separated into two one-electron processes, which suggests that the mixed-valence intermediates are stabilized by delocalized bonding. The high potential of
414Multiple Bonds Between Metal Atoms Chapter 9
the one-electron metal-based oxidation at ~ +0.85 V gives an indication of the greater stability of the Ru24+ core relative to that of the Ru25+ core in this environment.
Fig. 9.29. The structure of Ru2(meonp)4.
The Ru–Ru bond lengths of Ru24+ naphthyridines fall in the range 2.238-2.298 Å (Table 9.2), which is similar to those of the Ru24+ tetracarboxylates and tetraoxopyridinates. In addition, magnetic measurements conducted for the complexes [Ru2(O2CMe)3(bpnp)]PF6 and Ru2(meonp)4 show room temperature magnetic moments of 2.79 and 2.51 BM, respectively, which are consistent with the presence of two unpaired electrons.173,174 These structural and magnetic data give support to the μ2/4β2β*2/*2 electronic configuration.
9.4Ru26+ Compounds
Paddlewheel complexes having a Ru26+ core are relatively new additions to the family of Ru2 compounds. Other types of Ru26+ complexes that have been previously reported include compounds without bridging ligands, such as Ru2(dibenzotetraaza[14]annulene)(BF4)2,176 Ru2(CH2SiMe3)6,177 trihalo-bridged face-sharing bioctahedral compounds of the general type [Ru2X9]3-,178,179 and a variety of edge-sharing bioctahedral compounds with single-atom and three-atom bridging ligands.92,93 There are claims of paddlewheel Ru26+ carboxylate compounds,53,180 but such compounds do not exist.14 From the absence of an oxidation process in the cyclic voltammograms there would be no reason to expect these or any other [Ru2(O2CR)4]2+ species to be stable. However, with the use of either highly charged O,O'-donor, or electron rich bridging ligands in combination with suitable axial ligands, the higher oxidation state (Ru26+) becomes more favorable. The structurally characterized compounds of the Ru26+ core along with their corresponding Ru–Ru distances are given in Table 9.3.
Table 9.3. Structurally characterized Ru26+ paddlewheel compounds
Compound |
r(Ru–Ru) (Å) |
ref. |
O,O'-donor bridging ligands |
|
|
Cs2[Ru2(SO4)4(H2O)2] |
2.343(1) |
83 |
N,N'-donor bridging ligands |
|
|
aminopyridinate ligands |
|
|
(4,0)-Ru2(F5ap)4(C>CPh)2 |
2.441(1) |
110,183 |
(3,1)-Ru2(F5ap)4(C>CPh)2 |
2.475(1) |
110 |
trans-(2,2)-Ru2(F5ap)4(C>CPh)2 |
2.473(1) |
110 |

Ruthenium Compounds 415
Angaridis
Compound |
r(Ru–Ru) (Å) |
ref. |
|
(4,0)-Ru2(ap)4(C>CC>CSiMe3)2 |
2.472(1) |
114 |
|
(4,0)-Ru2(ap)4(CN)2·CH2Cl2·MeOH |
2.449[4] |
118 |
|
(3,1)-Ru2(2-Fap)4(CN)2·CH2Cl2 |
2.456(5) |
118 |
|
[(4,0)-Ru2(ap)4Cl][FeCl4]·2.5CH2Cl2 |
2.301(1) |
181 |
|
Ru2(F5ap)3(F4Oap)Cl·CH2Cl2·0.5benzene |
2.336(1) |
182 |
|
(4,0)-Ru2(ap)4(C>CPh)2 |
2.471(1) |
184 |
|
(4,0)-Ru2(ap)4(C>CPh)(C>CSiMe3) |
2.434(1) |
184 |
|
(4,0)-Ru2(ap)4(C>CSiPri3)(C>CC>CSiMe3) |
2.458(1) |
185 |
|
(4,0)-Ru2(ap)4(C>CC>CH)(C>CSiMe3) |
2.466(1) |
185 |
|
formamidinate ligands |
|
|
|
Ru2(DPhF)4(C>CPh)2 |
2.556[2] |
132 |
|
Ru2(DPhF)4(CN)2·2.5CH2Cl2·0.5hexane |
2.539(1) |
132 |
|
Ru2(DAnimF)4(C>CC>CSiMe3)2 |
2.599(1) |
135 |
|
Ru2(DPhF)4(C>CPh)2 |
2.556(1) |
187 |
|
Ru2(DPhp-ClF)4(C>CPh)2·2benzene |
2.555(1) |
188 |
|
(Me3SiC>CC>C)[Ru2(DPhF)4](µ-C>CC>CC>CC>C)[Ru2(DPhF)4]- |
2.559[2] |
189 |
|
(C>CC>CSiMe3)·4toluene·2hexane |
|||
|
|
||
[(But2bipy)(CO)3Re](py-4-C>C)[Ru2(DTolF)4](4-C>C-py)- |
2.567(1) |
190 |
|
[Re(CO)3(But2bipy)] |
|||
|
|
||
benzamidinate ligands |
|
|
|
Ru2(DMeBz)4Cl2·4THF |
2.323(1) |
191 |
|
Ru2(DMeBz)4(C>CSiMe3)2 |
2.450(1) |
191 |
|
Ru2(DMeBz)4(C>CC>CH)2 |
2.456(1) |
191 |
|
Ru2(DMeODMeBz)4Cl2·2CH2Cl2 |
2.316(1) |
192 |
|
Ru2(DEtBz)4Cl2 |
2.340(1) |
192 |
|
Ru2(DEtBz)4(C>CPh)2 |
2.459(1) |
192 |
|
Ru2(DEtBz)4(C>CSiMe3)2 |
2.461(1) |
192 |
|
Ru2(DMeODMeBz)4(C>CSiPri3)2 |
2.476(1) |
192 |
|
Ru2(m-MeODMeBz)4(C>CPh)2 |
2.448(1) |
193 |
|
Ru2(DMeBz)4(C>CC6H4-p-NO2)2·2CH2Cl2 |
2.459(1) |
193 |
|
[Ru2(DMeBz)4](BF4)2 |
2.265(1) |
194 |
|
[Ru2(DMeBz)4](NO3)2 |
2.287(1) |
194 |
|
other N,N'-donor bridging ligands |
|
|
|
[Ru2(dmat)4Cl]PF6 |
2.333(1) |
142 |
|
Ru2(hpp)4Cl2 |
2.321(1) |
195 |
9.4.1 Ru26+ compounds with O,O'-donor bridging ligands
The ability of “hard”, highly charged O,O'-donor bridging ligands, such as SO42- and -3+n, to stabilize the dimetal units in their higher oxidation states is well established.
Examples are the Mo25+ and Mo26+ complexes [Mo2(SO4)4]3- and [Mo2(HPO4)4(H2O)]2- (see Chapter 4). In 1989 the syntheses of the first Ru26+ compounds K2[Ru2(SO4)4(H2O)2] and Cs2[Ru2(SO4)4(H2O)2] and the crystal structure of the latter, which revealed a Ru–Ru bond length of 2.343(1) Å (Table 9.3), were reported.83, 84 The room temperature magnetic moments of both the K+ and Cs+ salts were ~4.5 BM which is in accordance with the presence of four unpaired electrons and the μ2/4β/*2β* electronic configuration.
A more convenient synthetic route for the above complexes utilizes K3[Ru2(O2CO)4]·4H2O as starting material to prepare the Ru25+ sulfate and phosphate compounds which are sub-

416Multiple Bonds Between Metal Atoms Chapter 9
sequently electrochemically oxidized to yield the corresponding Ru26+ compounds.82 Variable temperature magnetic susceptibility measurements for K2[Ru2(SO4)4(H2O)2] confirm the μ2/4β/*2β* electronic configuration. This is consistent with the Ru–Ru bond length of 2.343(1) Å in Cs2[Ru2(SO4)4(H2O)2], a distance that is slightly longer than that of 2.303(1) Å in the Ru25+ complex [Ru2(SO4)4(H2O)2]3- which is known to possess the μ2/4β2(/*β*)3 electronic configuration.83 The lengthening of the metal to metal distance on going from the Ru25+ to the Ru26+ complex is a combined effect of the loss of a β electron and the higher mean oxidation state which tends to weaken the Ru–Ru bonding interactions.
9.4.2 Ru26+ compounds with N,N'-donor bridging ligands
The possible existence of Ru26+ compounds with N,N'-donor bridging ligands had been originally suggested by electrochemical experiments on some Ru25+ compounds. A variety of Ru26+ compounds have been synthesized with the use of electron-rich N,N'-donor ligands, like aminopyridinates, formamidinates, benzamidinates and hpp, with or without the assistance of axial ligands.
Aminopyridinate ligands
Based on their structural characteristics Ru26+ tetraaminopyridinates can be divided into two groups: those without and those with axial ligands with μ donor and / acceptor ability. Examples of the former are [Ru2(ap)4Cl][FeCl4], [Ru2(ap)4F]PF6 and [Ru2(ap)4(H2O)2]CF3SO3. These are synthesized via simple oxidation reactions of Ru2(ap)4Cl with various oxidizing agents, such as Ag+ and [(δ5-C5H5)2Fe]+.181 Another compound, Ru2(F5ap)3(F4Oap)Cl, shown in Fig. 9.30, was synthesized serendipitously from the reaction of the (3,1) regioisomer of Ru2(F5ap)4Cl and a trace peroxide in THF.182 Only two complexes of this type have been characterized crystallographically, [Ru2(ap)4Cl][FeCl4] and Ru2(F5ap)3(F4Oap)Cl with Ru–Ru bond lengths of 2.301(1) and 2.336(1) Å, respectively (Table 9.3).
Fig. 9.30. The structure of Ru2(F5ap)3(F4Oap)Cl.
The room temperature magnetic moments of the above compounds are ~2.9 BM, which indicate the presence of two unpaired electrons.181 Thus, the ground state electronic configuration of these compounds can either be μ2/4β2/*2 or μ2/4β2/*1β*1. Structural data favor the former considering that the Ru–Ru bond length in [Ru2(ap)4Cl][FeCl4] is only 0.026 Å longer than the corresponding distance in Ru2(ap)4Cl, which has three unpaired electrons and the electronic configuration μ2/4β2(/*β*)3.96 Since the bond lengthening is so small, it is likely that the electron is removed from a β* molecular orbital upon oxidation, because removal of such an electron is expected to bring only a small shortening of the bond which is offset by