

Multiple Bonds Between Metal Atoms / 09-Ruthenium Compounds
.pdfRuthenium Compounds 417
Angaridis
an electrostatic repulsion between the Ru centers (higher mean oxidation state). On the other hand, removal of an electron from a /* molecular orbital would result in a substantial shortening of the Ru–Ru bond.
The second group of Ru26+ tetraaminopyridinates involves complexes with strongly bound μ donor and / acceptor ligands in axial positions, such as alkynyls and CN-. In a reinvestigation of the reactions between Ru25+ tetraaminopyridinates with excess of Li+ salts of alkynyls from which mono-alkynyl Ru25+ complexes are synthesized,112 both the mono-alkynyl and the bis-alkynyl Ru26+ tetraaminopyridinate complex Ru2(F5ap)4(C>CPh)2 were obtained in the reaction mixture and chromatographically separated.183 Other complexes of the type Ru2(Xap)4[(C>C)mY]2 (Y = H, Ph, SiMe3, SiPri3 and m = 1, 2) have been synthesized from similar reactions.114,184 An interesting extension is the synthesis of Ru26+ complexes with two different types of axially bound alkynyl ligands, such as Ru2(ap)4(C>CC>CH)(C>CSiMe3).185 Both the work-up conditions and the choice of the starting materials are crucial for the distribution of the products of these reactions. In the reaction of Ru2(ap)4Cl with Li(C>CC>CSiMe3) exposure of the reaction mixture to air is necessary in order to increase the yield of the bis-alkynyl complex Ru2(ap)4(C>CC>CSiMe3)2 (Fig. 9.31).114 The analogous reaction of Ru2(F5ap)4Cl with excess of Li(C>CPh) is more complicated, since Ru2(F5ap)4Cl exists as a mixture of the (4,0), (3,1) and trans-(2,2) regioisomers, and the product distribution depends on the type of isomer used as starting material, with the trans-(2,2) regioisomer giving the bis-acetylide compound as the only product.110
Fig. 9.31. The structure of Ru2(ap)4(C>CC>CSiMe3)2.
All the bis-alkynyl Ru26+ tetraaminopyridinates are isolated as airand moisture-stable solids which exhibit well resolved 1H NMR spectra. At least one very intense C>C stretching band is observed at ~2100 cm-1 in the IR spectra, while the analogous mono-alkynyl complexes and organic alkynyl compounds exhibit only weak C>C stretching bands. This could possibly be attributed to strong coupling between the two axial alkynyl ligands due to conjugation.115
The Ru26+ complexes with two axially coordinated CN- ligands, like Ru2(ap)4(CN)2 and Ru2(2-Meap)4(CN)2, are synthesized from the reactions of Ru25+ tetraaminopyridinates with excess of CN- and exposure of the reaction mixture to air.118 When Ru25+ tetraaminopyridinates with less basic ligands are used as starting materials, such as Ru2(2-Fap)4Cl, Ru2(2,4,6-F3ap)4Cl, Ru2(F5ap)4Cl, edge-sharing bioctahedral complexes of the type Ru2(µ- Fxap)2(δ2-Fxap)[µ-(o-NC)Fx-1ap](µ-CN) can also be isolated depending on the reaction conditions. For example, Ru2(2-Fap)4Cl reacts with excess of CN- at room temperature to give the dicyanide adduct and at 70 ºC to give Ru2(µ-2-Fap)2(δ2-2-Fap)[µ-(o-NC)ap]-(µ-CN),118 while Ru2(F5ap)4Cl gives only Ru2(µ-F5ap)2(δ2-F5ap)[µ-(o-NC)F4ap](µ-CN) (9.6a) and Ru2(µ- F5ap)2(δ2-F5ap)2(µ-CN)2 (9.6b).186
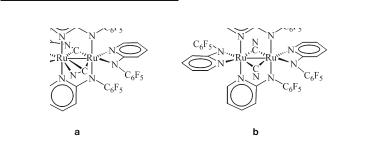
418Multiple Bonds Between Metal Atoms Chapter 9
9.6
Electrochemical studies of the bis-alkynyl and bis-cyano Ru26+ tetraaminopyridinates reveal three reversible, metal-centered, one-electron processes, one oxidation and two reductions which correspond to Ru27+, Ru25+ and Ru24+ complexes, respectively.110,118 These processes are irreversible for complexes with terminal C>CH groups.185 For the different regioisomers of Ru2(F5ap)4(C>CPh)2, an anodic shift for the oxidation and cathodic shifts for the reduction waves are observed proceeding from the (4,0) regioisomer to the (3,1) and to the trans-(2,2) regioisomer.110
The Ru–Ru bond lengths of the bis-alkynyl and bis-cyano Ru26+ tetraaminopyridinates fall in the range of 2.434 to 2.475 Å (Table 9.3). These are longer than the corresponding distances in the Ru26+ complex [Ru2(ap)4Cl][FeCl4] and the mono-alkynyl or the mono-cyano Ru25+ complexes. This difference can be attributed to the presence of the second axial alkynyl or cyano ligand, which further increases the antibonding μ* electron density between the two Ru atoms resulting in the lengthening of the Ru–Ru bond.
Considering that complexes of this type are diamagnetic (as it is shown by their normal 1H NMR spectra)110,118 and that the electronic configuration of the corresponding mono-alkynyl and mono-cyano Ru25+ tetraaminopyridinates is μ2/4β2(/*β*)3, the electronic configuration of μ2/4β2β*2 could be suggested for these Ru26+ compounds. However, this is not consistent with the observed long Ru–Ru bond lengths (2.434 to 2.475 Å), since the removal of an antibonding /* electron could not possibly lengthen the Ru–Ru bond from 2.33 to 2.47 Å. Because the Ru orbitals used to form axial bonds with μ donor ligands are also used to form the Ru–Ru μ bond, and because the alkynyl ligands are strong μ donors, these orbitals are deeply involved in the formation of the two axial Ru–C μ bonds with the alkynyl ligands. As a result, the Ru–Ru μ bond is essentially cancelled and the electron configuration of the metal-metal bonding orbitals becomes /4β2/*4. This leaves the Ru26+ core with a single net β bond, which explains satisfactorily the observed long Ru–Ru bond lengths.
Formamidinate ligands
The only type of Ru26+ tetraformamidinates known are those with two axially bound μ donor and / acceptor ligands. The first compound of this type, Ru2(DPhF)4(C>CPh)2, was synthesized from the reaction of Ru2(DPhF)4Cl with excess of Li(C>CPh).187 Others have been made similarly from reactions of Ru2(DArF)4Cl and excess of Li+ salts of CN-,132 or alkynyl ligands of the general type Y(C>C)m (m = 1, 2, and Y = Ph, SiMe3),135,188 followed by exposure of the reaction mixture to air and chromatographic purification which elutes only the bis-alkynyl Ru26+ complexes. Only one case has been reported in which both the monoand bis-alkynyl complexes can be separated by chromatography: these are the complexes Ru2(DPhF)4(C>CC>CSiMe3) and Ru2(DPhF)4(C>CC>CSiMe3)2.135 The formation of these types of complexes is influenced by the donor properties of the bridging formamidinate ligands; compounds with strong electron-

Ruthenium Compounds 419
Angaridis
withdrawing substituents form faster and they are isolated in higher yields than those with electron-donating substituents.188
Cyclic voltammetry studies show three reversible, one-electron, metal-centered processes, one oxidation and two reductions, corresponding to Ru26+ Α Ru27+ + e-, Ru26+ + e- Α Ru25+ and Ru25+ + e- Α Ru24+, respectively.188 The potential for each one of these processes depends on the substitution on the aryl groups of the formamidinate ligands. Linear correlations between these potentials and the substituent’s Hammett constants for a series of compounds of the type Ru2(DArF)4(C>CPh)2 have been established.
Complexes of this type are isolated as airand moisture-stable solids which are not thermally stable (most of them decompose above 50 ºC under vacuum) and they show normal 1H NMR spectra. Their IR spectra show one very intense band at ~2100 cm-1 corresponding to C>C stretching frequency, indicative of a strong coupling between the two axial alkynyl ligands due to the conjugation through the Ru26+ unit.115
Due to the rich electronic nature of Ru26+ units and the /-conjugation mediated by the alkynyl ligands, a variety of polymetallic Ru26+ alkynyl complexes have been synthesized and investigated as potential ‘molecular wires’. For example, (Me3SiC>CC>C)[Ru2(DPhF)4]- (µ-C>CC>CC>CC>C)[Ru2(DPhF)4](C>CC>CSiMe3) has a total length of ~3.5 nm and exhibits rich electrochemistry compared to that of the related complex Ru2(DPhF)4(C>CC>CSiMe3)2 complex.189 However, even though electronic delocalization occurs, the redox processes are not reversible. In contrast, the hetero-metallic complex [(But2bipy)(CO)3Re](py-4-C>C)- [Ru2(DTolF)4](4-C>C-py)[Re(CO)3(But2bipy)] displays electronic delocalization with reversible redox couples.190
The crystal structures of bis-alkynyl Ru26+ tetraformamidinates show deviations from the eclipsed configuration and distorted axial alkynyl ligands ( Ru–Ru–C ~160º), as shown in the structure of Ru2(DPhF)4(C>CPh)2 in Fig. 9.32. Based on theoretical calculations, it has been proposed that the origin of these distortions is electronic in nature and they have been attributed to a second-order Jahn-Teller effect.188
Fig. 9.32. The structure of Ru2(DPhF)4(C>CPh)2.
The Ru–Ru bond lengths fall in the range of 2.539 to 2.599 Å (Table 9.3). These distances are longer than those in the mono-alkynyl Ru25+ tetraformamidinates. The reason for this difference is the nature of the Ru2-alkynyl bonding interaction in the two types of compounds. In the mono-alkynyl Ru25+ tetraformamidinates the Ru25+-alkynyl bonding interaction is mainly a μ bonding interaction, but in the bis-alkynyl Ru26+ tetraformamidinates, it is a combination of μ bonding and d/-/* back-bonding interaction.188 As a result, not only the anti-bonding μ* electron density is increased, but also the / electron density is removed from the Ru26+ core

420Multiple Bonds Between Metal Atoms Chapter 9
resulting in the lengthening of the Ru–Ru bond. This gives a satisfactory explanation of the extremely elongated Ru–Ru bond of 2.599(1) Å observed in Ru2(DAnimF)4(C>CC>CSiMe3)2.135
The long Ru–Ru bond lengths of these compounds together with their diamagnetism (as indicated by their well resolved 1H NMR spectra) suggest that the electronic configuration is /4β2/*4. As in the case of the analogous Ru26+ aminopyridinates, the Ru–Ru μ bond is cancelled due to the formation of the Ru–C μ bonds with the strong μ donor alkynyl ligands. Theoretical calculations support this assignment and show that the Ru dz2 orbitals needed for the Ru–Ru μ bond are engaged in Ru–C μ bonding and μ* antibonding molecular orbitals, leaving the Ru26+ core with a net single β bond.188
Benzamidinate ligands
The first reported Ru26+ complex with bridging benzamidinate ligands, Ru2(DMeBz)4Cl2 (Fig. 9.33), was synthesized from the reaction of Ru2(O2CMe)4Cl with HDMeBz in the presence of Et3N and LiCl in THF.191 Two other complexes of this type, Ru2(DMeODMeBz)4Cl2 and Ru2(DEtBz)4Cl2, have been synthesized in a similar way.192 As discussed in section 9.2.3, the analogous reactions of Ru2(O2CMe)4Cl with formamidines give the corresponding tetraformamidinate compounds maintaining the Ru25+ core unoxidized. This difference can be attributed to the high basicity of the benzamidinate ligands which stabilizes higher oxidation states.
Fig. 9.33. The structure of Ru2(DMeBz)4Cl2.
The axial Cl- ions in the above complexes can be removed in reactions with excess of Li+ salts of alkynyl reagents to give bis-alkynyl Ru26+ tetrabenzamidinates.191-193 In addition, Ru2(DMeBz)4Cl2 reacts with AgBF4 and AgNO3 to yield complexes with weakly coordinating axial ligands, [Ru2(DMeBz)4](BF4)2 and [Ru2(DMeBz)4](NO3)2, respectively.194 These two axial chloride-free complexes offer an alternative route for the synthesis of bis-alkynyl Ru26+ tetrabenzamidinates under very mild reaction conditions.193
Cyclic voltammetry measurements of bis-chloro Ru26+ tetrabenzamidinates show three oneelectron, metal-based redox processes: a quasi-reversible oxidation, a reversible reduction and an irreversible reduction, which correspond to the formation of Ru27+, Ru25+ and Ru24+ complexes, respectively.191,192 Three redox processes are also observed in the electrochemistry of the complexes [Ru2(DMeBz)4](BF4)2 and [Ru2(DMeBz)4](NO3)2, which however are less reversible with anodically shifted potentials.194 The corresponding bis-alkynyl complexes exhibit similar redox behavior, but the redox waves are cathodically shifted due to the strong donating ability of the alkynyl ligands.191-193
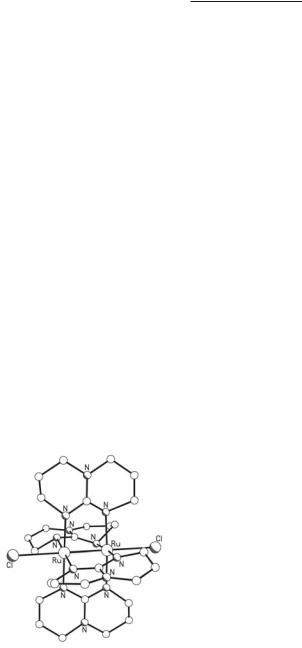
Ruthenium Compounds 421
Angaridis
Based on their Ru–Ru distances, which fall in the wide range of 2.265-2.476 Å as shown in Table 9.3, Ru26+ tetrabenzamidinates can be grouped in two categories. One category is formed by compounds with axial alkynyl ligands which exhibit long Ru–Ru distances that vary from 2.448 to 2.476 Å, while the other category contains complexes with axial Cl- ions or weakly coordinating BF4- and NO3- ions with much shorter Ru–Ru distances from 2.265 to 2.340 Å. The differences in the distances of the two types of compounds reflect their different electronic structures. In the complexes without axial alkynyl ligands the Ru–Ru bond lengths are similar to those observed in [Ru2(ap)4Cl][FeCl4]181 and Ru2(F5ap)3(F4Oap)Cl.182 Magnetic measurements show that they are paramagnetic with room temperature magnetic moments of ~3.0 BM which indicate the presence of two unpaired electrons.191,194 This is consistent either with the μ2/4β2/*2 or μ2/4β2/*1β*1 electronic configurations. By analogy to the Ru26+ aminopyridinates without axial alkynyl ligands, the observed Ru–Ru bond lengths favor the μ2/4β2/*2 electronic configuration. The bis-alkynyl Ru26+ tetrabenzamidinates display Ru–Ru bond lengths that are comparable to those of bis-alkynyl Ru26+ tetraaminopyridinates. In addition, they are diamagnetic, as indicated by their normal 1H NMR spectra.191 These data suggest that their electronic configuration is /4β2/*4. Similarly to the analogous Ru26+ tetraaminopyridinates, the formation of the Ru–C μ bonds with the strong μ donor alkynyl ligands cancels the formation of the Ru–Ru μ bond.
Other N,N'-donor bridging ligands
Two other N,N'-donor, electron rich ligands that are known to stabilize the dimetal units in high oxidation states are the guanidinate derivative hpp and dmat.
The complex Ru2(hpp)4Cl2 (Fig. 9.34) is synthesized by reacting Ru2(O2CMe)4Cl with an excess of molten Hhpp.195 The Ru–Ru bond length of 2.321(1) Å is very close to those of the bis-chloro Ru26+ tetrabenzamidinates, but much shorter than those of the corresponding diamagnetic Ru26+ complexes with axial alkynyl ligands (Table 9.3).
Fig. 9.34. The structure of Ru2(hpp)4Cl2.
Electrochemical studies show two one-electron, metal-centered redox processes: an oxidation at +0.55 V and a reduction at -0.60 V vs SCE, which correspond to Ru26+ Α Ru27+ + e- and Ru26+ + e- Α Ru25+, respectively. These are cathodically shifted with respect to the potentials of the bis-chloro Ru26+ tetrabenzamidinates191,192 and they suggest that hpp is more electron rich than benzamidinates. The mild oxidation potential of Ru2(hpp)4Cl2 implies that the oneelectron oxidized [Ru2(hpp)4Cl2]+ ion might be accessible; however, all attempts to generate it

422Multiple Bonds Between Metal Atoms Chapter 9
chemically or electrochemically have been unsuccessful and only decomposition products have been obtained.
The room temperature magnetic moment of Ru2(hpp)4Cl2 is 2.78 BM, which implies the presence of two unpaired electrons. Thus, the electronic configuration can either be μ2/4β2/*2 or μ2/4β2/*1β*1. No information can be obtained from EPR, since the complex is EPR silent. However, since Ru2(hpp)4Cl2 is isoelectronic with the Ru2(DMeBz)4Cl2 and the two complexes have similar Ru–Ru bond lengths, it is assumed that the electronic configuration is
μ2/4β2/*2.
The complex [Ru2(dmat)4Cl]PF6 is synthesized by bulk electrolysis of Ru2(dmat)4Cl in the presence of TBAPF6.142 The stabilization of the Ru26+ oxidation state is due to the high basicity of dmat, as indicated by its resonance structures in 9.7. Its room temperature magnetic moment is 2.89 BM, which suggests the presence of two unpaired electrons. Considering that the Ru–Ru bond length of 2.333(1) Å of this complex is similar to those observed in the Ru26+ tetraaminopyridinates without axial alkynyl ligands181,182 and the bis-chloro Ru26+ tetrabenzamidinates,191 the electronic configuration μ2/4β2/*2 has been proposed.
9.7
9.5Compounds with Macrocyclic Ligands
The only Ru2n+ compounds that have Ru–Ru bonds unsupported by bridging ligands are those of the type LRu–n RuL, where –n is a bond order of 2 to 3 and L represents a four-nitrogen macrocyclic ring. The earliest examples196 were the Ru2(tmtaa)2n (n = 0, +1, +2). From structural data197 and magnetic measurements, it was established that the electron configurations are μ2/4β2β*2/*2, μ2/4β2β*2/* and μ2/4β2β*2, respectively.
A series of compounds in which L is a porphyrin ligand (TPP2-, OEP2- or TPP2-) has also been prepared and studied.198-201 In terms of bonding, magnetism and structure these compounds differ little from the Ru2(tmtaa)2n species, but there are more of them and more extensive data. They react with neutral donors to give Ru(porph)L products.202,203
Mixed RuOs(porph)2 compounds have also been obtained,204,205 as have some (porph)RuMo(porph') and (porph)RuW(porph') compounds.205,206
With alkyl (Me, Et) substituted corroles (cor), the Ru26+ core is stabilized in (cor)Ru–Ru(cor) molecules,207 but reduction to Ru25+ and Ru24+, as well as oxidations to Ru27+ and Ru28+ compounds can be carried out electrochemically,208 although none of these oxidized or reduced species have been isolated.
9.6Applications
9.6.1 Catalytic activity
The study of catalytic activity of Ru2 compounds is limited to Ru2 tetracarboxylate complexes. Early studies using Ru2(O2CMe)4 and Ru2(O2CMe)4Cl showed that hydrogenation of alkenes and alkynes occurs in methanolic solution of fluoroboric acid in the presence of PPh3.209

Ruthenium Compounds 423
Angaridis
Since the active catalysts in these processes have not been isolated and characterized, the formation of a mononuclear Ru2+ complex that does the catalysis cannot be ruled out.
Room temperature hydrogenation of alk-1-ene by Ru2(O2CR)4 (R = CH3 or CF3) in the presence of 1 atm of H2 has also been reported.210 The suggested mechanism for this process is described by the following equations:
Ru2(O2CMe)4 + H2 Α HRu2(O2CMe)3 + H+ + CH3COO-
HRu2(O2CMe)3 + alkene Α Ru2(O2CMe)3(alkyl)
Ru2(O2CMe)3(alkyl) + H+ + CH3COO- Α Ru2(O2CMe)4 + alkane
No isomerization of the mono-alkenes is observed, suggesting irreversible alkyl formation. The slower rates of hydrogenation observed when the trifluoroacetate analog was used as catalyst are expected because of the slower H2 uptake by the electron poor dimetal core of Ru2(O2CCF3)4.
In addition, Ru2(O2CMe)4 has been found to catalyze the competitive cyclopropanation and cross-metathesis of alkenes.211 Small amounts of Ru2(O2CMe)4 added to a mixture containing styrene and norbornene together with ethyldiazoacetate (N2CHCO2Et) form cyclopropanated styrene and norbornene in 35-40% and 2% yields, respectively. The reaction is believed to be initiated by addition of N2CHCO2Et to Ru2(O2CMe)4 which results in the formation of a carbene species, Ru=CHCO2Et, that subsequently reacts with an olefin to form metallocyclobutane. This can facilitate metathesis or release of cyclopropane to give back the metal catalyst ready to react with another molecule of N2CHCO2Et.
Recently, Ru2(O2CMe)4 has been used as catalyst for the reaction of diazacoumarin with ROH at 90 ºC in alcohol or hexafluorobenzene as solvent to form 3-alkoxy-4-hydroxycoumarin, in yields of 35-95%, as a result of insertion into the O–H bond of the alcohols.212
9.6.2 Biological importance
The Ru25+ tetracarboxylate complexes have been used in antitumor activity studies. Ru2(O2CMe)4Cl and Ru2(O2CEt)4Cl show a small activity against P388 leukemia cells, but unfortunately the poor to moderate aqueous solubilities of these compounds did not allow tests with increased concentrations.213
In attempts to gain insight into the mechanism of antitumor activity, the binding of guanine bases to Ru25+ tetracarboxylate complexes has been studied. The compound [Ru2(O2CMe)2-x- (O2CCF3)x(9-EtGH)2(MeOH)2](O2CCF3)2·2MeOH·0.5Et2O (x = 0.18) (Fig. 9.35)214 is obtained by reacting Ru2(O2CMe)4Cl with AgO2CCF3 in refluxing CF3COOH and addition of two equivalents of 9-EtGH. It contains a Ru24+ core with the two metals separated by 2.322(13) Å and the two 9-EtGH groups in a cis head to tail (HT) fashion. The reactivity of Ru2(O2CMe)4Cl towards adenine and adenosine has also been studied.215 Although no crystal structures were reported, the products of the reactions were characterized by several physicochemical methods and were found to be a 1:1 adduct for adenine, which forms a polymeric chain with the adenine bridging the Ru25+ units through the axial positions, and a typical diadduct [Ru2(O2CMe)4- (adenosine)2]Cl for adenosine. The importance of these complexes, besides the binding of biologically relevant ligands, lies on their lesser toxicity compared to other diruthenium complexes, a property that allows the use of increased concentrations in antitumor activity tests.
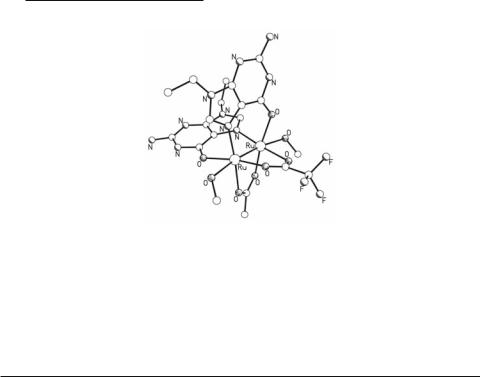
424Multiple Bonds Between Metal Atoms Chapter 9
Fig. 9.35. The structure of the cation in [Ru2(O2CMe)2-x(O2CCF3)x(9-EtGH)2(MeOH)2](O2CCF3)2 (x = 0.18).
A series of Ru25+ complexes of the type [Ru2(O2CR)4L2]+ (R = Me, CH=CH-Fc, m-C6H4SO3-, p-C6H4SO3-, L = Im, 1-MeIm, EtOH, H2O) having different water solubility and reduction potentials (Ru25+/Ru24+) was tested for anti-neoplastic activity against P388 leukemia cells.216 The compounds that showed significant activity were the water soluble m-C6H4SO3-, p-C6H4SO3- substituted complexes, and [Ru2(O2CMe)4(H2O)2]PF6. The others did not show any activity in the range of concentration used in the study due to their lack of water solubility.
References
1.T. A. Stephenson and G. Wilkinson, J. Inorg. Nucl. Chem. 1966, 28, 2285.
2.M. Mukaida, T. Nomura and T. Ishimori, Bull. Chem. Soc. Jpn. 1967, 40, 2462.
3.M. Mukaida, T. Nomura and T. Ishimori, Bull. Chem. Soc. Jpn. 1972, 45, 2143.
4.R. W. Mitchell, A. Spencer and G. Wilkinson, J. Chem. Soc., Dalton Trans. 1973, 846.
5.M. C. Barral, R. Jiménez-Aparicio, C. Rial, E. C. Royer, M. J. Saucedo and F. A. Urbanos, Polyhedron 1990, 9, 1723.
6.M. J. Bennet, K. G. Caulton and F. A. Cotton, Inorg. Chem. 1969, 8, 1.
7.A. Bino, F. A. Cotton and T. R. Felthouse, Inorg. Chem. 1979, 18, 2599.
8.M. C. Barral, R. Jiménez-Aparicio, D. Pérez-Quintanilla, E. Pinilla, J. L. Priego, E. C. Royer and
F.A. Urbanos, Polyhedron 1999, 18, 371.
9.T. Togano, M. Mukaida and T. Nomura, Bull. Chem. Soc. Jpn. 1980, 53, 2085.
10.B. K. Das and A. R. Chakravarty, Polyhedron 1991, 10, 491.
11.M. Abe, Y. Sasaki, T. Yamaguchi and T. Ito, Bull. Chem. Soc. Jpn. 1992, 65, 1585.
12.M. McCann, A. Carvill, P. Guinan, P. Higgins, J. Campbell, H, Ryan, M. Walsh, G. Ferguson and
J.Gallagher, Polyhedron 1991, 10, 2273.
13.T. Kimura, T. Sakurai, M. Shima, T. Togano, M. Mukaida and T. Nomura, Bull. Chem. Soc. Jpn. 1982, 55, 3927.
14.F. A. Cotton, M. Matusz and B. Zhong, Inorg. Chem. 1988, 27, 4368.
15.M. Spohn, J. Strähle and W. Hiller, Z. Naturforsch. 1986, 41b, 541.
16.M. McCann, E. Murphy, C. Cardin and M. Convery, Polyhedron 1993, 12, 1725.
17.F. A. Cotton, Y. Kim and T. Ren, Inorg. Chem. 1992, 31, 2723.
18.M. Handa, Y. Sayama, M. Mikuriya, R. Nukada, I. Hiromitsu and K. Kasuga, Chem. Lett. 1996, 201.
19.M. C. Barral, R. Jiménez-Aparicio, E. C. Royer, C. Ruíz-Valero, M. J. Saucedo and F. A. Urbanos,
Inorg. Chem. 1994, 33, 2692.

Ruthenium Compounds 425
Angaridis
20.M. C. Barral, R. Jiménez-Aparicio, J. L. Priego, E. C. Royer, M. J. Saucedo, F. A. Urbanos and
U.Amador, J. Chem. Soc., Dalton Trans. 1995, 2183.
21.M. C. Barral, R. Jiménez-Aparicio, J. L. Priego, E. C. Royer, F. A. Urbanos and U. Amador,
J.Chem. Soc., Dalton Trans. 1997, 863.
22.M. C. Barral, R González-Prieto, R. Jiménez-Aparicio, J. L. Priego, M. R. Torres and F. A. Urbanos,
Eur. J. Inorg. Chem. 2003, 2339.
23.K. D. Drysdale, E. J. Beck, T. S. Cameron, K. N. Robertson and M. A. S. Aquino, Inorg. Chim. Acta 1997, 256, 243.
24.M. C. Barral, R. Jiménez-Aparicio, E. C. Royer, C. Ruiz-Valero, F. A. Urbanos, E. GutiérrezPuebla and A. Monge, Polyhedron 1989, 8, 2571.
25.M. C. Barral, R. Jiménez-Aparicio, J. L. Priego, E. C. Royer, F. A. Urbanos and U. Amador, Inorg. Chem. 1998, 37, 1413.
26.G. G. Briand, M. W. Cooke, T. S. Cameron, H. M. Farrell, T. J. Burchell and M. A. S. Aquino,
Inorg. Chem. 2001, 40, 3267.
27.S. K. Mandal and A. R. Chakravarty, Inorg. Chim. Acta 1987, 132, 157.
28.B. K. Das and A. R. Chakravarty, Inorg. Chem. 1990, 29, 1783.
29.B. K. Das and A. R. Chakravarty, Inorg. Chem. 1990, 29, 2078.
30.B. K. Das and A. R. Chakravarty, Inorg. Chem. 1992, 31, 1395.
31.E. B. Boyar, P. A. Harding, S. D. Robinson and C. P. Brock, J. Chem. Soc., Dalton Trans. 1986, 1771.
32.F. A. Cotton, M. P. Diebold and M. Matusz, Polyhedron 1987, 6, 1131.
33.P. Stavropoulos, P. D. Savage, R. P. Tooze, G. Wilkinson, G.; B. Hussain, M. Motevalli and
M.B. Hursthouse, J. Chem. Soc., Dalton Trans. 1987, 557.
34.R. S. Drago, R. Cosmano and J. Telser, Inorg. Chem. 1984, 23, 4514.
35.M. C. Barral, R. Jiménez-Aparicio, J. L. Priego, E. C. Royer, E. Gutiérrez-Puebla and C. RuízValero, Polyhedron 1992, 11, 2209.
36.M. C. Barral, R. Jiménez-Aparicio, J. L. Priego, E. C. Royer, M. J. Saucedo, F. A. Urbanos and
U.Amador, Polyhedron 1995, 14, 2419.
37.F. A. Urbanos, M. C. Barral and R. Jiménez-Aparicio, Polyhedron 1988, 7, 2597.
38.M. W. Cooke, C. A. Murphy, T. S. Cameron, E. J. Beck, G. Vamvounis and M. A. S. Aquino,
Polyhedron 2002, 21, 1235.
39.H. J. Gilfoy, K. N. Robertson, T. S. Cameron and M. A. S. Aquino, Inorg. Chim. Acta 2002, 331, 330.
40.H. J. Gilfoy, K. N. Robertson, T. S. Cameron and M. A. S. Aquino, Acta Crystallogr. 2001, E57, m496.
41.G. Vamvounis, J. F. Caplan, T. S. Cameron, K. N. Robertson and M. A. S. Aquino, Inorg. Chim. Acta 2000, 304, 87.
42.H. Miyasaka, R. Clérac, C. S. Campos-Fernández and K. R. Dunbar, Inorg. Chem. 2001, 40, 1663.
43.D. Yoshioka, M. Handa, H. Azuma, M. Mikuriya, I. Hiromitsu and K. Kasuga, Mol. Cryst. Liq. Cryst. 2000, 342, 133.
44.Y. Liao, W. W. Shum and J. S. Miller, J. Am. Chem. Soc. 2002, 124, 9336.
45.D. Yoshioka, M. Mikuriya and M. Handa, Chem. Lett. 2002, 1044.
46.M. Handa, D. Yoshika, Y. Sayama, K. Shiomi, M. Mikuriya, I. Hiromitsu and K. Kasuga, Chem. Lett. 1999, 1033.
47.C. R. Wilson and H. Taube, Inorg. Chem. 1975, 14, 2276.
48.F. A. Cotton and E. Pedersen, Inorg. Chem. 1975, 14, 388.
49.M. McCann, A. Carvill, C. Cardin and M. Convery, Polyhedron 1993, 12, 1163.
50.M. W. Cooke, C. A. Murphy, T. S. Cameron, J. C. Swarts and M. A. S. Aquino, Inorg. Chem. Commun. 2000, 3, 721.
51.M. W. Cooke, T. S. Cameron, K. N. Robertson, J. C. Swarts and M. A. S. Aquino, Organometallics 2002, 21, 5962.
52.M. McCann and E. Murphy, Polyhedron 1992, 11, 2327.

426Multiple Bonds Between Metal Atoms Chapter 9
53.P. Higgins and G. M. McCann, J. Chem. Soc., Dalton Trans. 1988, 661.
54.J. E. Earley, R. N. Bose and B. H. Berrie, Inorg. Chem. 1983, 22, 1836.
55.A. O. Oyetunji, O. O. Olubuyide, J. F. Ojo and J. E. Earley, Polyhedron 1991, 10, 829.
56.M. Zhu, A. O. Oyetunji, K. Lu and J. E. Earley, Polyhedron 1989, 8, 577.
57.A. C. Dema and R. N. Bose, Inorg. Chem. 1989, 28, 2711.
58.M. Everhart and J. E. Earley, Polyhedron 1988, 7, 1393.
59.J. G. Norman Jr. and H. J. Kolari, J. Am. Chem. Soc. 1978, 100, 791.
60.J. G. Norman Jr., G. E. Renzoni and D. A. Case, J. Am. Chem. Soc. 1979, 101, 5256.
61.R. J. H. Clark and M. R. Franks, J. Chem. Soc., Dalton Trans. 1976, 1825.
62.D. S. Martin, R. A. Newman and L. M. Vlasnik, Inorg. Chem. 1980, 19, 3404.
63.R. J. H. Clark and L. H. Ferris, Inorg. Chem. 1981, 20, 2759.
64.V. M. Miskowski, T. M. Loehr and H. B. Gray, Inorg. Chem. 1987, 26, 1098.
65.V. M. Miskowski and H. B. Gray, Inorg. Chem. 1988, 27, 2501.
66.J. Telser and R. S. Drago, Inorg. Chem. 1984, 23, 3114.
67.F. A. Cotton, Y. Kim and T. Ren, Polyhedron 1993, 12, 607.
68.F. D. Cukiernik, D. Luneau, J.-C. Marchon and P. Maldivi, Inorg. Chem. 1998, 37, 3698.
69.R. Jiménez-Aparicio, F. A. Urbanos and J. M. Arrieta, Inorg. Chem. 2001, 40, 613.
70.F. D. Gukiernik, A.-M. Giroud-Godquin, P. Maldivi and J.-C. Marchon, Inorg. Chim. Acta 1994, 215, 203.
71.E. J. Beck, K. D. Drysdale, L. K. Thompson, L. Li, C. A. Murphy and M. A. S. Aquino, Inorg. Chim. Acta 1998, 279, 121.
72.M. C. Barral, R. Jiménez-Aparicio, D. Pérez-Quintanilla, J. L. Priego, E. C. Royer, M. R. Torres and F. A. Urbanos, Inorg. Chem. 2000, 39, 65.
73.M. Handa, Y. Sayama, M. Mikuriya, R. Nukada, I. Hiromitsu and K. Kasuga, Bull. Chem. Soc. Jpn. 1995, 68, 1647.
74.Y. Sayama, M. Handa, M. Mikuriya, I. Hiromitsu and K. Kasuga, Bull. Chem. Soc. Jpn. 2003, 76, 769.
75.Y. Sayama, M. Handa, M. Mikuriya, I. Hiromitsu and K. Kasuga, Chem. Lett. 1999, 453.
76.M. Handa, Y. Sayama, M. Mikuriya, R. Nukada, I. Hiromitsu and K. Kasuga, Bull. Chem. Soc. Jpn. 1998, 71, 119.
77.Y. Sayama, M. Handa, M. Mikuriya, I. Hiromitsu and K. Kasuga, Bull. Chem. Soc. Jpn. 2001, 74, 2129.
78.Y. Sayama, M. Handa, M. Mikuriya, I. Hiromitsu and K. Kasuga, Chem. Lett. 1998, 777.
79.Y. Sayama, M. Handa, M. Mikuriya, I. Hiromitsu and K. Kasuga, Bull. Chem. Soc. Jpn. 2000, 73, 2499.
80.A. J. Lindsay, G. Wilkinson, M. Motevalli and M. B. Hursthouse, J. Chem. Soc., Dalton Trans. 1987, 2723.
81.F. A. Cotton, L. Labella and M. Shang, Inorg. Chem. 1992, 31, 2385.
82.F. A. Cotton, T. Datta, L. Labella and M. Shang, Inorg. Chim. Acta 1993, 203, 55.
83.I. V. Kuz’menko, A. N. Zhilyaev, T. A. Fomina, M. A. Porai-Koshits and J. B. Baranovskii, Russ. J. Inorg. Chem. 1989, 34, 1457.
84.A. N. Zhilyaev, T. A. Fomina, I. V. Kuz’menko, A. V. Rotov and J. B. Baranovskii, Russ. J. Inorg. Chem. 1989, 34, 532.
85.X.-Y. Yi, L.-M. Zheng, W. Xu and S. Feng, Inorg. Chem. 2003, 42, 2827.
86.T. Malinsky, D. Chang, F. N. Feldmann, J. L. Bear and K. M. Kadish, Inorg. Chem. 1983, 22, 3225.
87.A. R. Chakravarty and F. A. Cotton, Polyhedron 1985, 4, 1957.
88.A. R. Chakravarty, F. A. Cotton and D. A. Tocher, Polyhedron 1985, 4, 1097.
89.M. C. Barral, R. Jiménez-Aparicio, J. L. Priego, E. C. Royer, F. A. Urbanos, A. Monge and C. RuízValero, Inorg. Chim. Acta 1993, 12, 2947.
90.M. C. Barral, I. de la Fuente, R. Jiménez-Aparicio, J. L. Priego, M. R. Torres and F. A. Urbanos,
Polyhedron 2001, 20, 2537.