

Хохлов
.pdfГлава 1. Теоретические основы создания лекарственного средства
Исследование сифилиса, финансируемое Службой общественного здравоохранения США в городе Таскиги – это еще один вопиющий пример нарушения этических принципов проведения клинических исследований с участием человека. «Эксперимент, целью которого было исследование всех стадий сифилиса, длился в период с 1932 года по 1972. В начале эксперимента сформировали две группы: 399 носителей заболевания и 201-го здорового человека как контрольная группа» [486]. Участникам эксперимента не предлагалось стандартного метода лечения сифилиса пенициллином. За больными участниками велся контроль и наблюдение с целью предотвращения их обращения за лечением в другие клиники, не вовлеченные в эксперимент. К концу такого исследования естественного течения болезни из 399человекзараженныхсифилисомвживыхосталосьлишь74.28человека умерли от сифилиса, 100 – от осложнений, вызванных им. Было заражено 40 их жен и 19 детей, родившихся с врожденным сифилисом [217].
С 1940 года Комиссия по атомной энергии США спонсировала проведение экспериментов на людях, ставших невольными и неосведомленными участниками таких экспериментов [337]. Людям, попадавшим в больницы по разным причинам, тайно вводили 4,7 микрограммов плутония. Все участники думали, что это часть их лечения. Эксперимент не ограничивался только больными и был распространен на детей-сирот и заключенных в штатах Орегон и Вашингтон. Целью такого эксперимента стало установление степени действия радиоактивного материала на человеческий организм, для дальнейшей оценки использования радиации в целях защиты или нападения во времена Холодной войны [232].
Все приведенные случаи явилась серьезным стимулом для развития законодательства в области клинических исследований.
В настоящее время рациональное конструирование эффективных лекарств для этиотропной или патогенетической терапии становится возможным благодаря прорывам в молекулярной биологии, геномике, компьютерных технологиях.
Обычно на создание нового лекарства исследователей толкает так называемая неудовлетворенная потребность: если против какого-либо заболеваниянетэффективныхпрепаратовлибосуществующиелекарстванедостаточно эффективны или вызывают тяжелые побочные эффекты. Стимулом может стать появление новых технологий, открытия в этиологии и патогенезе заболеваний. Например, сочетание неудовлетворенной потребности с появлением новых технологий в последние годы привело к появлению многих новых противоопухолевых препаратов.
Онкология – одна из самых показательных сфер медицины в плане неудовлетворенной потребности. Зачастую злокачественные опухоли диагностируются на поздних стадиях, когда лечение может носить лишь паллиативный характер.
Одновременно за последние десятилетия сильно шагнули вперед технологии. Многие открытия сделаны в сфере молекулярной генетики. Ученые
11

Промышленная фармация. Путь создания продукта
узнали о молекулярных механизмах, которые способствуют развитию и прогрессированию рака, улучшилось понимание работы противоопухолевого иммунитета.
Этодалотолчоккпоявлениюсразудвухперспективныхнаправленийвлечении онкологических заболеваний: таргетной терапии и иммунотерапии.
В1986 году два исследователя – Стэнли Коэн и Рита Леви-Монтальчини
–получили Нобелевскую премию за открытие факторов роста. В дальнейшем это сыграло колоссальную роль в развитии онкологии. Оказалось, что повышенная активность трансмембранных белков из семейства рецепторныхтирозинкиназ(EGFR,HER2)играетважнуюрольвразвитииипрогрессировании рака.
Мишень была найдена, и вскоре появились способные поражать ее препараты. В 2002 году в Японии одобрили первый таргетный препарат, способный ингибировать «неправильный» EGFR - гефитиниб («Иресса»). В 2003 году его одобрило и FDA. Сегодня у врачей уже есть большой выбор ингибиторов факторов роста и других «молекул-мишеней».
Вреалиях современного мира есть еще два фактора, влияющие на принятие решения о разработке новых лекарственных препаратов: коммерческая целесообразность и политические интересы. Яркий пример – вакцина против вируса Зика. Правительства разных стран пытаются бороться с этой проблемой. Например, на территории Европейского Союза предусмотрена система наград и стимулов для компаний, разрабатывающих педиатрические препараты.
Другая стратегия предполагает движение «от потенциального препарата». Есть вещество, обладающее свойствами, которые могут быть полезны
вмедицине. Задача – найти точки приложения. Один из примеров недавно обнаруженного потенциального препарата – соединение в составе яда пау-
ка Hadronyche infensa.
Когда ученые проанализировали яд паука Hadronyche infensa, оказалось,
что в нем содержится белок Hi1a. Дальнейший анализ показал, что этот белок блокирует ионные каналыASIC1a, работа которых играет важную роль в повреждении клеток головного мозга при инсульте. В ходе экспериментов на лабораторных мышах Hi1a помог предотвратить повреждение мозга при остром нарушении мозгового кровообращения, восстановить неврологические функции. Результаты исследования опубликовали в апреле 2017 года, и есть надежда, что на основе «паучьего» белка удастся создать новые препараты.
Есть и третья «стратегия» – непредсказуемая. Иногда лекарства создают случайно. Пожалуй, самый показательный случай – тот, что произошел с ученым, не любившим наводить порядок в лаборатории, Александром Флемингом. 3 сентября 1928 года, вернувшись из отпуска, микробиолог заметил, что в некоторых чашках со стафилококками выросли плесневые грибы, а сами бактерии погибли. Так был открыт пенициллин, и наступила эра антибиотиков [136].
12

Глава 1. Теоретические основы создания лекарственного средства
1.3. Этапы жизненного цикла лекарственных препаратов
1.3.1. Краткий обзор основных этапов жизненного цикла лекарственного препарата: отличия российской и зарубежной регуляторной практик
Термин «жизненный цикл лекарственного препарата» введен Международным советом (конференцией) по гармонизации технических требований, предъявляемых к регистрации лекарственных препаратов для медицинского применения (ICH) в 2005 г. В руководстве ICH Q8 Pharmaceutical development (Фармацевтическая разработка) жизненный цикл лекарственного препарата определяется как «все фазы жизни препарата от начальной разработки до его реализации и окончательного вывода из оборота» [346].
В общеэкономическом смысле лекарственный препарат можно рассматривать как товар. Концепция жизненного цикла продукта предложена Т. Левиттомв1965г.[397].Онаописываетпериодвремени,втечениекоторого товар обращается на рынке, с позиций объема продаж: внедрение на рынок, рост, зрелость и спад (уход с рынка). Однако на фармацевтический рынок влияют не только сугубо экономические факторы, социально-экономиче- ский и демографический состав населения, и даже не структура заболеваемости, т.е. спрос в чистом виде. Специфика лекарственного препарата как товара отражается на существенных особенностях его жизненного цикла.
Принципиальными являются следующие отличия фармацевтической продукции [214]:
–меньшее влияние конечного потребителя на выбор приобретаемого лекарственного препарата, в особенности, отпускаемого по рецепту, и, тем более, инновационного препарата в отсутствии возможности генерической замены;
–меньшая зависимость спроса на лекарственный препарат от изменения цены на него;
–невозможность оценить качество продукта и его потребительские свойства (полезность, бесполезность или вред) ни потребителем перед приобретением, ни врачом перед рекомендацией препарата в каждом конкретном случае;
–невозможность возврата средств / замены товара / компенсации вреда здоровью в случае неэффективности или непереносимости препарата.
Помимо товарной специфики лекарства, его предназначение для использования в лечебном процессе, в интересах жизни и здоровья пациента, объясняет более жесткий характер требований, предъявляемых к качеству фармацевтической продукции в сравнении с прочими товарами массового потребления. При этом выборочный контроль качества путем разрушения препарата, по определению, не может охватить 100% продукции. Данное обстоятельство обусловило формирование системы обеспечения и контроля качества на всех этапах жизненного цикла препарата – так называемых,
13

Промышленная фармация. Путь создания продукта
надлежащих практик, GxP, имеющих мировое признание. В частности, руководством ICH Q8 подчеркивается, что качество не может быть полностью проверено на этапе финального контроля; качество необходимо заложить при разработке и обеспечить при производстве [346]. Примечательно, что в отличие от классического определения жизненного цикла продукта, который традиционно оценивается с момента выведения товара на рынок, в определении жизненного цикла лекарственного препарата подчеркивается роль начальных этапов разработки.
Фармацевтический рынок, кроме того, отличает беспрецедентная зарегулированность. Главная цель широкого вмешательства государства в сферу обращения лекарственных средств – обеспечение качества, эффективности и безопасности фармацевтической продукции [84]. К регуляторным функциям государства, осуществляемым органами исполнительной власти разного уровня, согласно Федеральному закону от 12.04.2010 N 61-ФЗ «Об обращении лекарственных средств» (далее 61-ФЗ) [106] относятся, в частности:
–лицензирование производства лекарственных средств и фармацевтической деятельности (в т. ч. импорта, сбыта);
–организация экспертизы лекарственных средств, этической экспертизы возможности проведения клинического исследования лекарственного препарата, экспертизы документов, представленных для определения возможности рассматривать лекарственный препарат при государственной регистрации в качестве орфанного;
–организация проведения комплексной оценки лекарственного препарата в целях принятия решений о возможности включения лекарственного препарата в перечень жизненно необходимых и важнейших лекарственных препаратов (ЖНВЛП);
–выдача разрешений на проведение клинических исследований лекарственных препаратов;
–государственная регистрация лекарственных препаратов;
–организация и (или) проведение инспектирования субъектов обращениялекарственныхсредствнасоответствиеправиламнадлежащихпрактик;
–государственная регистрация установленных производителями лекарственных препаратов предельных отпускных цен на ЖНВЛП;
–установление предельных размеров оптовых надбавок и предельных размеров розничных надбавок к фактическим отпускным ценам, установленным производителями лекарственных препаратов, на лекарственные препараты, включенные в перечень ЖНВЛП;
–осуществление государственного контроля за применением цен на лекарственные препараты, включенные в перечень ЖНВЛП, организациями оптовой торговли, аптечными организациями, индивидуальными предпринимателями, имеющими лицензию на фармацевтическую деятельность;
–установление порядка ввоза и вывоза лекарственных средств в РФ и вывоза лекарственных средств из РФ;
–осуществление фармаконадзора;
14

Глава 1. Теоретические основы создания лекарственного средства
–установление порядка формирования регистрационного досье на лекарственный препарат и требований к документам в его составе;
–утверждение правил рационального выбора наименований лекарственных препаратов для медицинского применения;
–утверждение перечня наименований лекарственных форм;
–утверждение требований к инструкции по медицинскому применению лекарственных препаратов.
В истории немало примеров успешных с экономической точки зрения лекарственных препаратов, у которых при выявлении новых рисков для пациентов (т.е. изменения соотношении «польза – риск» в неблагоприятную сторону) регулятором принималось решение по отмене регистрации препарата, либо производителем проводилось добровольное изъятие препарата из обращения (Биопарокс, Катадолон, Меридиа, Эреспал и др.). Таким образом, исключительно экономический подход к делению жизненного цикла товара на этапы является недостаточно информативным для описания жизненного цикла лекарственного препарата [58].
Жизненный цикл лекарственного препарата во многом зависит от его типа с регуляторной точки зрения и, соответственно, требований, предъявляемых к регистрационному досье инновационного (нового, оригинального, референтного), воспроизведенного, гибридного, биоаналогового препаратов.
Наиболее долгий путь от открытия до потребителя у инновационного (оригинального) лекарственного препарата. Согласно определению Всемирной организации здравоохранения (ВОЗ), инновационный фармацевтический продукт – это продукт, впервые зарегистрированный на рынке на основании документации о его качестве, безопасности и эффективности [414]. Правилами регистрации и экспертизы лекарственных средств для медицинского применения, действующими на территории Евразийского экономического союза (ЕАЭС), предусмотрено понятие «оригинального лекарственного препарата», по сути, являющегося эквивалентом определения инновационного лекарственного препарата, данного ВОЗ. Согласно формулировке, оригинальный лекарственный препарат – это лекарственный препарат с новым действующим веществом, который был первым зарегистрирован и размещен на мировом фармацевтическом рынке на основании досье, содержащего результаты полных доклинических (неклинических) и клинических исследований, подтверждающих его качество, безопасность и эффективность [104]. Следует заметить, что в регуляторной терминологии США и ЕС термин «оригинальный» лекарственный препарат не используется. Данные препараты определяются как «новые лекарства» или препараты, регистрируемые на основании раздела 505(b) Федерального закона о пищевых продуктах, лекарствах и косметике в США [281]. В ЕС они называются лекарственными препаратами, регистрируемыми на основании самостоятельного досье, либо регистрируемыми в соответствии с положениями статьи 8(3) Директивы 2001/83/EC [273].
15

Промышленная фармация. Путь создания продукта
Основными этапами жизненного цикла нового (инновационного, оригинального, первого в классе) препарата являются следующие:
−разработка и исследования (научно-исследовательские и опытно-кон-
структорские разработки, НИОКР; Research & Development, R&D);
−поиск (Screening);
−фармацевтическая разработка (Pharmaceutical development);
−доклинические исследования (Preclinical study);
−клинические исследования (Clinical trials);
−государственная регистрация (Registration; в ЕС – лицензирование,
Мarketing authorisation);
−производство (Manufacturing);
−хранение (Storage);
−сбыт (Marketing);
−лонч (запуск, выведение на рынок, Launch);
−оптовая реализация (Wholesale);
−розничная реализация (Retail sales);
−медицинское применение (Medical use);
− пострегистрационный мониторинг эффективности и безопасно-
сти лекарственного препарата (Postapproval / Postmarketing surveillance & Pharmacovigilance);
−утилизация (Disposal);
−вывод из обращения (Product Discontinuation).
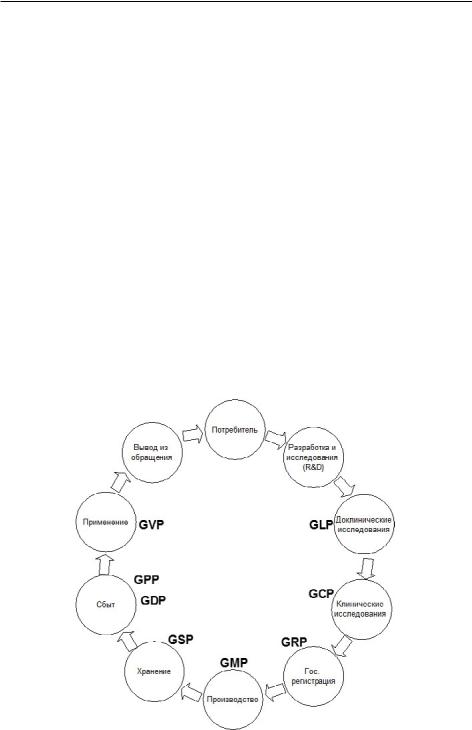
Как упомянуто выше, в целях обеспечения качества лекарственного препаратакаждыйэтапегожизненногоцикластандартизируетсянадлежащими практиками, GxP. Имеющие мировое признание руководства по надлежащим практикам представляют собой не детальные документы, а обобщение главных принципов и требований, предъявляемых к разработке, исследованию, производству и реализации лекарственных средств. Среди руководств GxPможно выделить основные:
−надлежащая лабораторная практика (Good Laboratory Practice, GLP);
−надлежащая клиническая практика (Good Clinical Practice, GCP);
−надлежащая регуляторная практика (Good Regulatory Practice, GRP);
−надлежащая фармакопейная практика (Good Pharmacopoeial Practice, GPhP);
−надлежащая производственная практика (Good Manufacturing Practice, GMP);
−надлежащая практика фармаконадзора (Good Pharmacovigilance Practices, GVP);
−надлежащая практика хранения лекарственных средств (Good Storage Practice, GSP);
−надлежащая дистрибьютерская практика (Good Distribution Practice, GDP);
−надлежащая аптечная практика (Good Pharmacy Practice, GPP);
−надлежащая инженерная практика (Good Engineering Practice, GEP)
−надлежащая тканевая практика (Good Tissue Practice, GTP) и др.
16
Глава 1. Теоретические основы создания лекарственного средства
Арсенал надлежащих практик непрерывно пополняется. За последнее время разработаны, в частности, GCLP (Good Clinical Laboratory Practice надлежащая практика клинических лабораторий), GEP (Good Epidemiology Practice надлежащая эпидемиологическая практика), GPeP (Good Pharmacoepidemiology Practice, надлежащая фармакоэпидемиологическая практика), GCDMP (Good Clinical Data Management Practice, надлежа-
щая практика управления клиническими данными), GAMP (Good Automated ManufacturingPractice,надлежащаяпрактикаавтоматизированногопроизвод-
ства), GACP (Good Agriculture and Collection Practice, надлежащая практика выращивания,сбораихраненияисходногосырьярастительногопроисхождения),GPP(GoodProcurementPractice,надлежащаяпрактиказакупки),атакже GPP (Good Publication Practice, надлежащая практика публикаций), надлежащая хроматографическая практика (Good Chromatography Practices) и др.
Таким образом, система GxP создает замкнутую цепь менеджмента качества, звенья которой последовательно охватывают все стадии жизненного цикла лекарственного препарата (Рис. 1). Наряду с надлежащими практиками, в фармации и медицине используется целый ряд международных согласительных документов и национальных стандартов, отраслевых и ведомственных приказов в сфере разработки, исследований и рационального применения лекарственных препаратов, реализуемых на соответствующих стадиях жизненного цикла. К таким документам, в частности, относится система руководств ICH (см. далее).
Рис. 1. Схема жизненного цикла инновационного лекарственного препарата и имплементации основных надлежащих практик на соответствующих этапах [58] (с изм.).
17

Промышленная фармация. Путь создания продукта
В 61-ФЗ дефиниция, передающая суть «инновационности» или «оригинальности» лекарственного препарата отсутствует. Используется понятие «референтного»препарата.Вместестемивнаучнойлитературе,ивофициальных документах РФ термин «инновационное лекарственное средство» имеет широкое хождение, а разработка таких препаратов отечественными предприятиями поддерживается государственной программой «Развитие фармацевтической и медицинской промышленности» [108]. В настоящее время рассматривается законопроект «О внесении изменений в Федеральный закон «Об обращении лекарственных средств», в рамках которого вводится понятие оригинального лекарственного препарата
Под референтным лекарственным препаратом в 61-ФЗ понимается лекарственный препарат, впервые зарегистрированный в РФ, качество, эффективность и безопасность которого доказаны на основании результатов доклинических исследований лекарственных средств и клинических исследований лекарственных препаратов, проведенных в соответствии с требованиями частей 6 и 7 статьи 18 закона в отношении лекарственных средств для медицинского применения <…>, и который используется для оценки биоэквивалентности или терапевтической эквивалентности, качества, эффективности и безопасности воспроизведенного или биоаналогового (биоподобного) лекарственного препарата [106]. Как следует из определения, референтным лекарственным препаратом не обязательно является новый, оригинальный, т. н. первый в классе (first-in-class) препарат. Им может стать любой, в т. ч. воспроизведенный, препарат, отвечающий требованиям законодательства РФ и впервые зарегистрированный на территории страны. Законом разрешается использование такого препарата в качестве эталона в исследованиях биоэквивалентности.
Концепция референтности в регуляторных документах ЕАЭС и ЕС сосредоточена на функции сопоставления. Под референтным понимается лекарственный препарат, используемый в качестве препарата сравнения и являющийся эталоном, по которому определяются (нормируются) свойства лекарственного препарата [104, 276]. Очевидно, что в таком контексте оригинальность референтного препарата не является необходимым условием
– все зависит от цели и дизайна конкретного исследования. Так, в исследовании биоэквивалентности воспроизведенных, биоаналогичных, гибридных препаратов референтным будет являться оригинальный лекарственный препарат. В исследованиях с дизайном non-inferiority / equality / superiority (доказательство не меньшей / одинаковой / превосходящей эффективности или безопасности) референтный препарат не обязательно должен быть оригинальным и может даже относиться к другому классу лекарственных средств, с иным механизмом действия. При изменении процесса производства биоподобного препарата референтным будет биоаналог, произведенный до внесения изменений [98].
Для референтного лекарственного препарата, произведенного и зарегистрированного за рубежом, с учетом требования об обязательности клинических исследований всех препаратов на этапе регистрации (кроме ряда исключений), жизненный цикл непосредственно в РФ начинается с проведения клинических исследований II-III фаз. Согласно ст. 38 п. 1.1. 61-ФЗ,
18
Глава 1. Теоретические основы создания лекарственного средства
установление безопасности лекарственных препаратов для здоровых добровольцев и (или) переносимости их здоровыми добровольцами (т.е. клинические исследования I фазы) дополнительно не проводится, если такие исследования произведены за пределами РФ [106] (см. раздел 3.7).
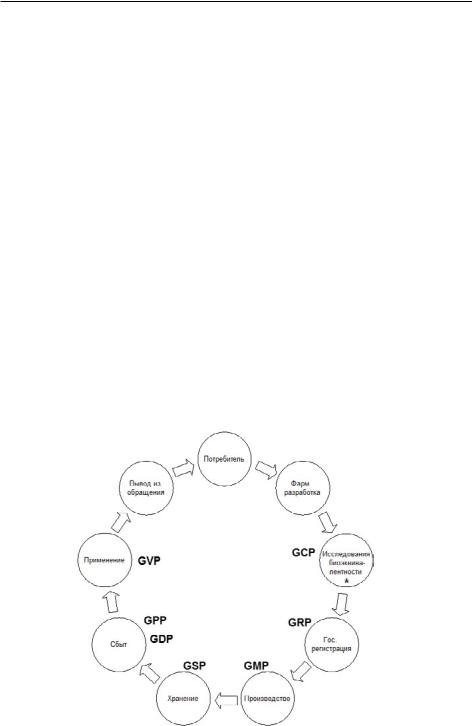
Воспроизведенный лекарственный препарат по сравнению с оригинальным имеет более короткий и менее затратный путь к потребителю (Рис. 2). (определение воспроизведенного препарата см. раздел 3.8). Определение формально совпадает с определением воспроизведенного препарата (дженерика),даннымэкспертамиEMA[276]иЕАЭС[104].Внекоторыхстранах полностьюповторятьпрограммуисследованияоригинальногопрепаратане требуется, и в большинстве случаев достаточно доказательства биоэквивалентности воспроизведенного препарата референтному.
В Европейской Директиве 2001/83/EC под референтным препаратом в контексте исследований биоэквивалентности подразумевается оригинальный препарат (зарегистрированный в соответствии со статьей 893) [273]. В формулировках нормативной документации ЕАЭС воспроизведенный препарат в одном случае соотносится с оригинальным [104], в другом – с референтным без уточнения регистрационного статуса последнего.
Согласно правилам проведения исследований биоэквивалентности, принятыми в ЕАЭС [111] и ЕС [320], изучение биодоступности in vivo при соблюдении необходимых условий не требуется:
−для дополнительных дозировок (биовейвер),
−для отдельных видов лекарственных форм,
−для процедуры биовейвер, основанной на биофармацевтической системе классификации.
Рис. 2. Схема жизненного цикла воспроизведенного лекарственного препарата Примечание: * – в ряде случаев не требуется (см. текст).
19

Промышленная фармация. Путь создания продукта
Часто дженерик называется «копией оригинала». В действительности же воспроизведенный препарат представляет собой результат собственной фармацевтическойразработкисдопустимымиотличиямиоторигинальногопрепарата. Например, в рамках законодательства РФ, ЕАЭС, ЕС фармацевтическая альтернативность в отношении активных субстанций (различные соли, эфиры, комплексы, изомеры, кристаллические формы и другие производные одного и того же действующего вещества) признается незначимой, если безопасность и (или) эффективность воспроизведенного и референтного препарата значимо не отличаются [106]. Кроме того, различные лекарственные формы для приема внутрь с немедленным высвобождением признаются в рамках исследований биодоступности одной и той же лекарственной формой. Также допускаются различия в составе вспомогательных веществ, не значимые для терапевтической сопоставимости и биоэквивалентности воспроизведенного и референтного лекарственного препарата. Однако если доказательства сопоставимой эффективности и безопасности в случае фармацевтических альтернатив или различий в составе вспомогательных веществ у воспроизведенного и референтного препаратов потребуют проведения дополнительных исследований, данный «воспроизведенный» препарат с регуляторной точки зрения будет уже рассматриваться как гибридный.
Понятие «гибридного лекарственного препарата», не подпадающего под определение воспроизведенного лекарственного препарата при невозможности подтверждения его биоэквивалентности с помощью исследований биодоступности, а также в случае, если в данном препарате произошли изменения действующего вещества (веществ), показаний к применению, дозировки, лекарственной формы или пути введения по сравнению с оригинальным препаратом, предусмотрено нормативной базой ЕАЭС и ЕС [104].
Концепция гибридности, аналогично концепции оригинальности и воспроизведенности, является исключительно регуляторной и характеризует объемпрограммыисследований,которыйявляетсядостаточнымдляподачи заявления о регистрации лекарственного препарата. Как и в случае воспроизведенного, гибридный препарат похож на оригинальный (референтный). Активная часть молекулы действующего вещества, содержащаяся в самом препарате или образующаяся в организме в результате биотрансформации, будет одинаковой у воспроизведенного (гибридного) и оригинального препарата. Однако если с помощью традиционного исследования биодоступности подтвердить биоэквивалентность воспроизведенного препарата оригинальному невозможно (например, в отношении препарата, оказывающего местное действие, применяемого местно или наружно), либо данного исследования недостаточно для полноценной характеристики эффективности и безопасности модифицированного лекарственного препарата (в т. ч. при изменении действующего вещества, лекарственной формы, дозировки, путивведения,показанийкприменению)длярегистрациитакогопрепарата потребуются дополнительные доклинические или клинические исследования (Рис. 3). В этом случае регистрационное досье будет смешанным, а пре-
20