

Хохлов
.pdfГлава 2. Прогнозирование свойств фармакологических веществ in silico
гут быть получены при использовании методов, комбинирующих подходы молекулярной механики, молекулярной динамики и квантовой химии [468], что требует применения суперкомпьютеров для выполнения соответствующих расчетов [180, 181].
Критическое рассмотрение приближений и ограничений существующих в настоящее время методов докинга приведено в работе [257]. Проблемы с применением этих методов, обычно связаны с недостаточным качеством информации о 3D структуре макромолекул-мишеней, сложностями учета разнообразия конформационных состояний гибких лигандов, приближенностьюрасчетаоценокаффинностисвязывания.Этипроблемыобязательно должны приниматься во внимание при интерпретации результатов виртуального скрининга с применением докинга.
2.4.4. Дизайн веществ на основе структуры лигандов
Дизайн лекарств на основе структуры-лигандов (Ligand-Based Drug Design) исторически является первым рациональным подходом к поиску фармакологически активных веществ. Анализ эмпирических результатов тестирования биологической активности различных веществ в процессе фармакологических и токсикологических исследований позволил сформулировать заключение, что «сходные вещества вызывают сходные эффекты». На этой основе уже в середине XIX века предпринимаются попытки связать химическую структуру и физико-химические свойства веществ с их биологической активностью. В 1848 г. J. Blake предположил, что биологическая активность соли определяется ее основным или кислотным компонентом, а не солью в целом (отметим, что теория электролитической диссоциации была предложена Аррениусом только в 1884 г.). В середине XIX века Brown и Fraser показали, что некоторые конвульсанты меняют свою активность и превращаются в миорелаксанты, когда третичный атом азота в их молекуле при метилировании становится четвертичным. Ричардсон при исследовании влияния различных функциональных групп на биологическую активность пришел к выводу, что существует некий общий закон, связывающий структуру с активностью. В 1869 году он писал: «Я уверен, что наступит время, когда книги, называемые «фармакопеи», будут построены на основе рассуждений,икогдахимикиврачбудутдействоватьзаодно»(цит.поработе[140]).
В 2019 году отмечается 150-летие открытой Д.И. Менделеевым Периодической системы элементов, согласно которой: «Свойства простых тел, а также формы и свойства соединений элементов, а потому и свойства образуемых ими простых и сложных тел, стоят в периодической зависимости от их атомного веса». Установленный Д.И. Менделеевым закон можно рассматривать как первый пример анализа глобальных взаимосвязей «структу- ра-свойство», позволивший предсказать существование шести ранее неизвестных элементов и довольно точно рассчитать их свойства [141].
H. Meyer (1899) и E. Overton (1901) выдвинули «липидную теорию кле-
точной депрессии», объясняющую разную выраженность эффекта депрес-
61

Промышленная фармация. Путь создания продукта
сантов их различным сродством к липидам клеток центральной нервной системы, определяемым по значению коэффициента распределения между липидным растворителем и водой (цит. по [10]).
Начало современного этапа анализа связи «структура-активность» датируется 60-ми годами XX века. Связано оно с работами Ханша [328, 331], Фри и Вильсона [292] и Кира [374], которые положили начало для математизации, а позднее – компьютеризации этих разработок.
Подход, предложенный Ханшем и соавторами, основывается на предположении о влиянии различных свойств молекулы (коэффициент распределения октанол-вода, электронные и стерические факторы) на ее взаимодействие с биологическим объектом. Для многих классов химических соединений и различных видов активности удалось представить эту QSAR (Quantitative Structure-Activity Relationships) зависимость в унифицирован-
ном виде:
где: P – коэффициент распределения вещества между водной и липидной фазами (например, октанол-вода), характеризующий гидрофобность вещества; G – константа Гамета-Тафта, характеризующая электронные свойства; E – стерический параметр.
Несомненным преимуществом данного уравнения является наличие вполне определенного «физически обусловленного» механизма взаимодействия вещества с биологическим объектом, и возможность расчета независимых параметров на основе простой аддитивной схемы по константам заместителей, которые могут быть взяты из соответствующих таблиц.
С подходом Ханша связана целая эпоха в фармацевтических разработках. Во многих случаях на этой основе удалось построить корреляционные уравнения, характеризующие связь структуры веществ одного химического ряда с конкретной биологической активностью. В то же время, немало примеров, когда статистически значимые зависимости не были найдены [330].
Согласно предложенной Фри и Вильсоном аддитивной модели предполагается, что биологическая активность может быть описана в виде суммы эффектов заместителей плюс некоторая постоянная величина, отражающая вклад в активность общей структурной основы членов данного гомологического ряда:
где:cij –вкладвактивностьi-гозаместителя,находящегосявструктуревj-м положении,такчтоcij =1,еслиэтоимеетместо,иcij =0впротивномслучае. Коэффициенты kij находят с помощью линейного регрессионного анализа. Преимуществом данной модели является то, что для построения соответствующей регрессионной зависимости необходима только информация о химической структуре и биологической активности веществ, включенных в обучающую выборку. Недостатком – жесткая привязка к набору заместителей, содержащихся в соединениях обучающей выборки, что, в ряде слу-
62

Глава 2. Прогнозирование свойств фармакологических веществ in silico
чаев, не позволяет осуществить прогноз для новых веществ. В ряде работ были предложены модификации данного метода, детальное сопоставление которых было проведено Кубиньи и Керханом [386–388]. Показано, что эта модель эквивалентна модели Ханша, содержащей только члены первого порядка. Несмотря на сравнительную простоту этого подхода, а может быть именно благодаря ей, различные модификации метода Фри-Вильсона применяются в анализе связи «структура-активность» до настоящего времени (см., например, работу [434]).
Кир использовал методы теоретического конформационного анализа для расчета устойчивых конформаций соединений обучающей выборки. Далее производилсяпоискфрагментов,имеющихсходноераспределениезарядов, что позволило осуществить «картирование» активного центра. С использованием этого подхода Кир построил двумерные модели активных центров ацетилхолина, серотонина, гистамина, стероидов и др. [373, 374].
Около 70 примеров успешного применения QSAR к веществам из различных химических классов и разнообразным видам биологической активности приведено в книге [291]. Одним из примеров такого рода является построение регрессионных уравнений, описывающих взаимосвязь между ингибированием моноаминоксидазы и физико-химическими свойствами пропиниламинов [407]. На основе этих уравнений был выполнен прогноз величины биологической активности (pI50 – отрицательный логарифм молярной концентрации, при которой происходит ингибирование 50% фермента) для шести веществ, не включенных в выборку, по которой строились регрессионные уравнения. Для соединений (52–56) из данной работы экспериментальные величины pI50 составили 5, 5; 4, 0; 3, 7; 3, 7; 3, 0; 2, 9, а расчетные 6, 1; 4, 1; 4, 4; 3, 6; 2, 4; 2, 9, соответственно [407], что свидетельствуето достаточновысокомкачествепредсказаний. Следует отметить, что рассмотренные в книге R. Franke виды биологической активности преимущественно относятся к надмолекулярному уровню (антибактериальная, противогрибковая, антималярийная, цитостатическая, спазмолитическая, антигипертензивная, анальгетическая, и др.), что отражает преобладающие в тот период методики экспериментального тестирования биологической активности.
В настоящее время (Q)SAR модели – классификационные или количественные модели, широко используются в фармацевтических исследованиях и разработках для прогнозирования биологической активности (в том числе взаимодействия с мишенями) химических соединений. Традиционно, классификационное SAR моделирование применяют для разделения соединений на активные и неактивные по отношению к определенному свойству молекул (например, антигипертензивное, канцерогенное, мутагенное, анксиолитическое действие и т.д.). QSAR модели применяют для оценки количественных величин активности (например, IC50). Модели строят на основе данных о биологической активности химических соединений, полученных экспериментально. В начале 2000-х годов были разработаны критерии
63

Промышленная фармация. Путь создания продукта
(OECD принципы, Organisation for Economic Co-operation and Development) построения QSAR моделей (OECD (Q)SAR project) [425]:
1.Четко определенное свойство, для которого создается QSAR модель.
2.Однозначный алгоритм построения зависимости «структура-актив- ность».
3.Определенная область применимости.
4.Четкие критерии оценки надежности прогноза и предсказательной способности.
5.Механистическая интерпретация (соответствие реальным механизмам прогнозируемых взаимодействий, если это возможно).
Применение (Q)SAR моделей в поиске новых фармакологических веществ основано на том, что фармакологические эффекты конкретного соединения, вызываемые его взаимодействием с молекулярной мишенью, зависят от особенностей его структуры. Следовательно, описав структуру соединения набором характерных элементов (молекулярных дескрипторов)
ипроанализировав ряд соединений, обладающих одинаковыми свойствами, на основании схожести их структурной организации можно построить зависимость, вероятнее всего прогнозирующую проявление конкретного свойства. Построенная зависимость может быть использована для оценки наличия величины данного свойства для новых соединений и отбора «кандидатов», согласно прогнозу, обладающих данным свойством.
Таким образом, необходимыми элементами при построении (Q)SAR зависимостей являются дескрипторное описание химической структуры, представление биологической активности, математический метод для анализа взаимосвязей «структура-активность», наличие обучающей выборки.
Внастоящее время разработано несколько тысяч различных структурных дескрипторов, включая структурные фрагменты, топологические индексы, различные физико-химические параметры, квантово-химические характеристики, и др. [489].
Представление биологической активности может быть качественным либо количественным. В первом случае, фиксируется факт наличия или отсутствия активности («активно/неактивно», «1/0»), и для анализа взаимосвязей структура-активность используются классификационные модели, которые позволяют отнести новое соединение к категории «активных» или «неактивных» с оценкой соответствующих вероятностей. Во втором случае для оценки величины активности используют количественные параметры, характеризующие «выраженность» активности, такие как IC50, EC50, CC50,
идр., которые рассматриваются в качестве независимых переменных при построении количественных зависимостей QSAR, например, с использованием регрессионных моделей.
Для оценки взаимосвязей «структура-активность» используют самые разные математические подходы, включая метод ближайших соседей, искусственные нейронные сети, деревья решений, множественную линейную регрессию, метод опорных векторов, Байесовскую статистику, глубокое ма-
шинное обучение, и др.
64

Глава 2. Прогнозирование свойств фармакологических веществ in silico
Для создания (Q)SAR модели, достоверно отражающей реальные процессы взаимодействия между фармакологической мишенью и химическим соединением, требуется репрезентативная обучающая выборка. Скорость накопления данных об известных взаимодействиях для различных классов соединений многократно увеличилась после внедрения технологии высокопроизводительного скрининга (High-Throughput Screening, HTS) как инструмента прикладных или академических исследований [380]. Параллельно с увеличением скорости накопления данных появляются электронные ресурсы, агрегирующие получаемую информацию и предоставляющие к ней доступ. Полученные данные проходят фильтрацию на основе их качества и возможности использования для моделирования [294, 360, 478]. От тщательности подготовки обучающей выборки непосредственно зависит качество построенных моделей и, соответственно, шансы на успех при использовании этих моделей для поиска новых фармакологических веществ и оптимизации их характеристик [491]. Более детальная информация о современном состоянии исследований в области (Q)SAR представлена в уже упоминавшейся выше монографии О.А. Раевского [141].
2.5. Прогноз на основе анализа структурного сходства
Как уже говорилось выше, методы дизайна лекарств, основанные на структуре лигандов, исходят из предположения, что «Similar molecules exert similar biological activities» (схожие молекулы обладают схожей биологической активностью) [384]. В подавляющем большинстве случаев эта эмпирическая гипотеза находит экспериментальное подтверждение, что широко используется в фармацевтической химии для конструирования новых аналогов известных фармакологических веществ.
В частности, именно по этой причине защита интеллектуальной собственности в области фармацевтики, как правило, осуществляется в виде «зонтичных патентов», нередко содержащих структурные формулы тысяч схожих по структуре химических веществ, представленных в виде формулы Маркуша [193]. При этом экспериментальные результаты исследования биологической активности представлены в патенте для отдельных «примеров», а для остальных молекул наличие соответствующей активности предполагается априори без проведения тестирования.
На основе предположения об аналогичном фармакологическом действии структурно схожих молекул выполнены многочисленные работы в области медицинской химии, направленные на поиск и оптимизацию свойств новых биологически активных веществ [508]. Использование информации об активности/свойствах «ближайших структурных соседей» позволяет повысить точность (Q)SAR/(Q)SPR моделей [141]. Это предположение широко применяется с целью получения оценок токсикометрических характеристик химических веществ на основе так называемого “ReadAcross” подхода [459].
65

Промышленная фармация. Путь создания продукта
В то же время, известны случаи, когда структурно сходные молекулы обладают различной биологической активностью [34, 384]. При рассмотрении общего пространства химико-биологических взаимодействий выделяют так называемые “activity cliffs” [271]. Эти случаи могут представлять особый интересдляпоискаоригинальныхлекарственныхвеществвмалоисследованных областях химико-биологического пространства и, напротив, являются серьезной проблемой при построении и применении (Q)SAR моделей [266].
Известно, что не существует универсального метода оценки структурного сходства, пригодного «на все случаи жизни» [243, 462]. С этой целью используютсамыеразныедескрипторыхимическойструктурыиразличные количественные характеристики, характеризующие сходство. В литературе даже обсуждается вопрос, действительно ли существуют “activity cliffs”, или же это артефакты, обусловленные несовершенством используемых расчетных методов [336, 408].
Тем не менее, оценки структурного сходства широко используют как для поиска структурных аналогов в химических базах данных, таких как
ChemNavigator [252], или Integrity [260]. Так, например, в базе данных
ChemNavigator, введя в качестве запроса структурную формулу ацетилсалициловой кислоты, исследователь получит «на выходе» структурные формулы содержащихся в этой базе данных аналогов этой лекарственной суб-
станции (Рис. 9).
Рис. 9. Поиск структурных аналогов ацетилсалициловой кислоты в базе данных ChemNavigator (представлены три наиболее похожие структуры)
66

Глава 2. Прогнозирование свойств фармакологических веществ in silico
Как видно из рисунка 9, помимо структурных формул доступных аналогов, исследователю предоставляются: идентификатор структуры (ID) в базе данных ChemNavigator, молекулярный вес (MW), расчетное значение логарифма коэффициента распределения октанол-вода (cLogP), а также принадлежность к конкретной библиотеке (Collection), показатель чистоты (Purity) и (в некоторых случаях) ориентировочный срок поставки образца (Shipping Window).
Оценка величины сходства в простейшем случае осуществляется на основе так называемого коэффициента Танимото (TC). Для двух структурных формул А и B коэффициент Танимото может быть рассчитан по формуле:
где:c –числосовпадающихструктурныхэлементовуанализируемыхмоле- кул; a – число структурных элементов у молекулыA; b – число структурных элементов у молекулы B.
В качестве структурных элементов могут использоваться различные дескрипторы, например, фрагменты. При расчете сходства удобным представлением структуры является битовая строка, в которой «1» означает наличие соответствующего дескриптора в молекуле, «0» – его отсутствие. Значения коэффициента Танимото находятся в интервале между нулем и единицей, либо могут быть выражены в процентах. Необходимо подчеркнуть, что в некоторых случаях величина TC=100% не гарантирует идентичности структурных формул сравниваемых молекул – возможно, что совпадающие структурные элементы по-разному объединены в структуре молекулAи B.
Можно предполагать, что в случае химических соединений, имеющих достаточно сложную структуру, оценки активности/свойства химического соединениянаосновеструктурногосходствамолекулбудутменееточными, чем на основе (Q)SAR зависимостей или моделей фармакофоров, поскольку специфическое взаимодействие лиганда с рецептором обеспечивается отдельными структурными элементами лиганда, а структурное сходство рассчитывают для всей молекулы целиком. В работе [406] на основе анализа нескольких выборок фармакологических веществ из библиотеки фирмы Abbottбылопоказано,чтопризначенииTC>85%лишьв30%случаевмолекулы, действительно, обладают сходными свойствами.
Тем не менее, во многих случаях поиск аналогов по сходству является «методомвыбора»припоискеновыхфармакологическихвеществ,поскольку, в особенности для представляющих особый интерес новых молекулярных мишеней, количество известных лигандов на ранних стадиях исследований составляет от одной до нескольких молекул, что недостаточно для построения (Q)SAR зависимостей или моделей фармакофоров.
67

Промышленная фармация. Путь создания продукта
2.6.Построение моделей фармакофоров
Вотличиеотпрогноза«посходству»,когдавкачестве«образца»(pattern) используетсяинформацияоструктурнойформулеоднойактивноймолекулы (в случаеприменения метода «k ближайших соседей»происходитпопарное сравнение каждой из молекул с известными свойствами с новой молекулой
споследующей интеграцией полученных оценок), при построении модели фармакофора осуществляется суперпозиция набора молекул, что позволяет установить структурные элементы, необходимые для наличия активности.
Согласно Глоссарию [60], «Фармакофор, фармакофорная модель (pharmacophore; pharmacophoric pattern) – совокупность стерических и электрон-
ных особенностей, необходимых для обеспечения оптимальных надмолекулярных взаимодействий лиганда со специфической биологической мишенью и для стимулирования (или блокирования) биологического ответа. Фармакофор не является реальной молекулой или реальным сочетанием функциональных групп. Это чисто абстрактная концепция, которая позволяет описать общие возможности межмолекулярных взаимодействий для группы соединений по отношению к их мишени. Фармакофор можно рассматривать как наибольшую общую часть (максимально перекрывающиеся части) всего набора активных молекул».
Таким образом, при построении модели фармакофора «отсекаются» структурные элементы лиганда, которые представляются несущественными для взаимодействия «мишень-лиганд», что позволяет снизить «информационный шум», возникающий при расчете оценки попарного сходства структурных формул молекул.
На рис. 10 приведен пример построения модели фармакофора для ингибиторов моноаминоксидазы (МАО) А [30]. На основе структурных формул ингибиторов этого фермента из двух различных химических классов производные оксазолидинонов и пиразинокарбазолов), были определены подструктуры, существенные для проявления активности; пересечение этих подструктур позволило получить объединенный фармакофор для ингибиторов МАО А. Аналогичным образом была построена модель фармакофора для ингибиторов МАО В. Сопоставление этих двух фармакофорных моделей, приведено на рис. 11; видны существенные различия структурных требований к ингибиторам этих ферментов. Использование построенных фармакофорных моделей позволяет осуществлять виртуальный скрининг или конструирование селективных ингибиторов по отношению к каждому из этих ферментов.
Взависимости от доступной информации построение моделей фармакофоров может осуществляться с использованием 2D или 3D подходов. В первом случае, модель фармакофора учитывает только топологическую связность в молекулах, что не позволяет, учесть различия в биологической активности энантиомеров (например, упоминавшийся выше талидомид состоит из двух энантиомеров R и S, из которых только S-энантиомер про-
68

Глава 2. Прогнозирование свойств фармакологических веществ in silico
являет тератогенное действие [411]) или пространственное расположение функциональных групп относительно друг друга. Моделирование пространственной структуры лигандов позволяет принять во внимание различия в активности различных энантиомеров.
Рис. 10. Построение модели фармакофора для ингибиторов МАО А
Рис. 11. Фармакофорные модели для ингибиторов МАО А (А) и МАО В (В)
Популярнымметодом3DQSARявляетсяCoMFA(ComparativeMolecular FieldAnalysis – сравнительный анализ молекулярных полей) [278]. Условием применимости данного метода является наличие выборки соединений, одинаковым образом взаимодействующих с одним и тем же рецептором (одинитотжесайтсвязывания,однаитажегеометриярасположенияфункционально важных групп). На первом этапе осуществляют суперпозицию всех молекул в трехмерном пространстве, что можно сделать с помощью модели фармакофора. На следующем этапе вокруг этих молекул строится решетка, в узлах которой рассчитывают энергию взаимодействия пробного атома, помещенного в каждый узел (атом углерода, положительно или отрицательно заряженный атом, донор или акцептор водородной связи) с каждой молекулой. В результате дескрипторами в CoMFAвыступают величи-
69
Промышленная фармация. Путь создания продукта
ны энергии взаимодействия пробного атома и набором молекул. Далее методом частичных наименьших квадратов устанавливают корреляции между рассчитанными значениями энергий и значениями биологической активности [141]. Близким по идеологии является метод CoMSIA (Comparative Molecular Similarity Index Analysis – сравнительный анализ индексов моле-
кулярного подобия) [378].
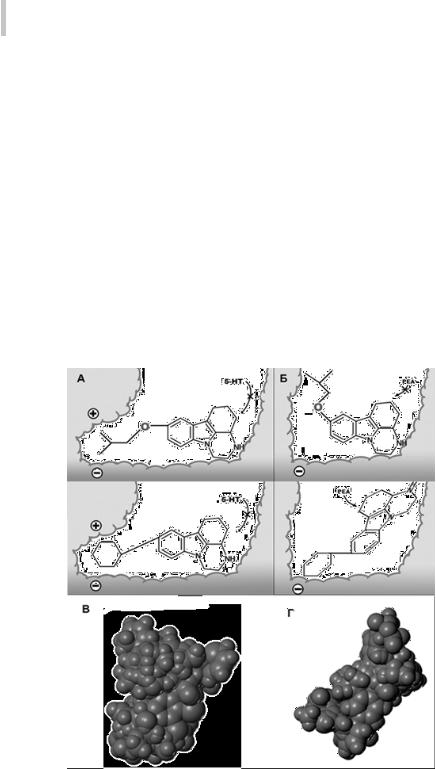
С применением 3D QSAR и CoMFAна основе анализа выборок ингибиторов МАО А и МАО В производных пиразинкарбазола, индола, изатина и карбобензоксиэтиламина были построены модели активных центров МАО А и МАО В (рис. 12). Сопоставление с расшифрованными впоследствии трехмерными структурами обоих ферментов показало достаточно хорошее соответствие. С использованием построенных моделей были выявлены селективные и неселективные ингибиторы МАО А и МАО В [29].
Наряду с уже упоминавшейся выше возможностью дифференциации взаимосвязей «структура-активность» для различных энантиомеров, преимуществом использования 3D методов для построения моделей фармакофоров является получение непосредственной информации о возможных расстояниях между отдельными фармакофорными точками в пространстве. Для обеспече-
Ри. 12. Модели активных центров и их слепков для МАО А (А и В) и МАО В (Б и Г)
70