

Хохлов
.pdfГлава 1. Теоретические основы создания лекарственного средства
фекта лекарственного средства», размещенной на интернет-сайте Росздравнадзора.
2. Дистанционно через Интернет напрямую в подсистему «Фармаконадзор» АИС Росздравнадзора. В настоящее время этот способ рассматривается как предпочтительный и более удобный.
По результатам мониторинга Федеральным законом №61-ФЗ предусмотрена возможность приостановления применения ЛП и даже отмена государственной регистрации.
Оборот конкретного лекарственного препарата может быть приостановлен решением Росздравнадзора в случаях:
−когда получены информация о наличии не указанных в инструкции к лекарственному препарату побочных и нежелательных реакций;
−когда выявлены особенности взаимодействия препаратов друг с другом, а также серьезные побочные эффекты, которые представляют опасность для жизни и здоровья человека;
−когда инструкции лекарственного препарата не соответствуют его эффективности и безопасности.
Решения Минздрава России публикуются на сайте grls.rosminzdrav.ru. На официальном сайте Росздравнадзора также доступна информация об изменении профиля безопасности ЛС, представленная в соответствующих информационных письмах.
Фармаконадзор осуществляется на основании:
––сообщений субъектов обращения лекарственных средств; ––периодических отчетов по безопасности лекарственных препаратов,
направляемых в Росздравнадзор держателями или владельцами регистрационных удостоверений лекарственных препаратов или уполномоченными ими иными юридическими лицами;
––периодическихотчетовпобезопасностиразрабатываемого(исследуемо- го) лекарственного препарата, направляемых в Росздравнадзор юридическими лицами, на имя которых выданы разрешения на проведение клинических исследованийвРФ,либодругимиуполномоченнымиюридическимилицами; ––информации, полученной в ходеосуществления государственного кон-
троля (надзора) в сфере обращения лекарственных средств.
ПорядокрасширяеттребованиякпредставлениювРосздравнадзорсрочной отчетности о нежелательных реакциях и периодической отчетности по безопасности лекарственных препаратов. Устанавливаются требования к согласованию планов управления рисками, предоставляемыми держателями регистрационных удостоверений в Росздравнадзор, при выявлении проблем безопасности лекарственных средств.
Также документом регламентируется деятельность Росздравнадзора по анализу поступающих данных о безопасности лекарственных средств и подготовке рекомендаций Минздраву России по ограничению обращения лекарственных препаратов в случае, если риск их применения превышает возможную пользу.
41

Промышленная фармация. Путь создания продукта
Исполнение производителями и разработчиками лекарственных препаратов требований Порядка делает необходимым наличие на предприятиях работающей системы фармаконадзора.
Результатом деятельности Федеральной службы по надзору в сфере здравоохранения по развитию государственной системы мониторинга безопасности лекарственных препаратов является рост числа и улучшение качества поступающих сообщений о нежелательных реакциях. По сравнению с показателями 2011 года в 2014 году сообщаемость о нежелательных реакциях лекарственных препаратов увеличилась на 42%. Анализ сообщений, поступивших в АИС Росздравнадзора за 2011–2014 годы, показал, что наиболеераспространенныминежелательнымиреакциямиприприменении лекарственных препаратов являются аллергические (24, 8% сообщений). В основном развитие аллергических реакций было обусловлено применением антибактериальных препаратов цефалоспоринового, пенициллинового и фторхинолонового рядов.
Втечение указанного периода несколько возросло количество сообщений
онедостаточной терапевтической эффективности лекарственных препаратов (13, 4% от общего числа сообщений). В целом структура распределения нежелательных реакций по ведущей клинической симптоматике соответствует данным отечественной и зарубежной научной литературы. Среди фармакотерапевтических групп за 2011–2014 годы по количеству сообщений лидируют антимикробные препараты (29, 6%) и лекарственные препараты, влияющие на сердечно-сосудистую систему (19, 3%). В значительной степени это обусловлено активным мониторингом нежелательных реакций на антикоагулянтные препараты, проводимым компаниями-производителями.
Следует сказать, что количество компаний-производителей, ведущих исследования и мониторинг нежелательных реакций на выпускаемые лекарственные препараты, в последнее время выросло, и этот факт, конечно, поспособствовал росту числа сообщений (по контрацептивным препаратам, антикоагулянтам, противоопухолевым средствам, препаратам для терапии ожирения).
Всего в 2016 г. в Автоматизированную информационную систему (АИС) Росздравнадзора поступило и рассмотрено 27, 5 тыс. сообщений.
Следует отметить, что Российская Федерация, наряду со всеми странами, в настоящее время входящими в Таможенный союз, с 1998 г. является официальным участником международной программы ВОЗ по мониторингу безопасности лекарственных средств, поэтому многие аспекты деятельности по выявлению, анализу и оценке безопасности ЛП в нашей стране проходят постоянный процесс гармонизации с международными стандартами фармаконадзора, вырабатываются единые правила мониторинга НР в рамках стран Таможенного союза.
Совет Евразийской экономической комиссии 3 ноября 2016 года принял ряд документов, направленных на создание системы общего рынка лекарственных средств на территории Евразийского экономического союза. Сре-
42

Глава 1. Теоретические основы создания лекарственного средства
ди них было принято Решение № 87 «Об утверждении Правил надлежащей практики фармаконадзора Евразийского экономического союза».
Данное решение устанавливает единые требования к системе качества фармаконадзора, к организации деятельности уполномоченных органов го- сударств-членов ЕАЭС в сфере фармаконадзора, устанавливает обязанности держателей регистрационных удостоверений по мониторингу данных по фармаконадзору, оценке безопасности ЛП, подготовке периодических отчетов о безопасности, контролю качества системы фармаконадзора, назначению уполномоченных лиц в сфере фармаконадзора, формированию и поддержанию мастер-файла системы фармаконадзора, предоставлению сведений в уполномоченный орган, и т.д.
Решение также устанавливает порядок проведения инспекций системы фармаконадзора, организации независимого аудита системы фармаконадзора, порядок сотрудничества государств-членов ЕАЭС в сфере обмена информацией по инспекциям системы фармаконадзора и иные требования. Согласно Решению, нормы Правил, установленные в отношении инспектирования системы фармаконадзора, вступили в силу с 01.01.2017.
1.3.8. Прекращение производства
Всоответствии с Решением Совета Евразийской экономической комиссии от 03.11.2016 №77 «Об утверждении Правил надлежащей производственной практики Евразийского экономического союза» целью мероприятий по прекращению производства является эффективное управление конечным этапом жизненного цикла продукции. Для управления мероприятиямипопрекращениюпроизводствадолжныиспользоватьсяпредварительно установленные подходы:
––хранение документации и образцов,
––постоянная оценка продукции (работа с претензиями, испытания стабильности),
––составление отчетности в соответствии с установленными требованиями.
На данном этапе жизненного цикла должны также применяться все элементы системы фармацевтической системы качества.
Мониторинг эффективности процесса и качества продукции (такой как испытания стабильности) должен продолжаться до завершения всех испытаний.Всоответствиисустановленнымитребованиями,испытаниянеобходимо продолжать на препаратах, находящихся в обороте.
Всистеме корректирующих и предупреждающих мероприятий должно учитываться влияние на продукцию, которая остается в обороте, а также на продукцию, на которую могло быть оказано влияние.
Любые изменения после прекращения выпуска продукции должны проходить через соответствующую систему управления изменениями.
Проверка со стороны руководства должна включать такие элементы, как стабильностьпродукцииирасследованиепретензийвотношенииеекачества.
43

Промышленная фармация. Путь создания продукта
Глава 2. ПРОГНОЗИРОВАНИЕ СВОЙСТВ ФАРМАКОЛОГИЧЕСКИХ ВЕЩЕСТВ
IN SILICO
2.1. Предпосылки для поиска новых лекарственных препаратов
Создание нового лекарственного препарата основывается на анализе обширного массива разнородной информации, накопленной человечеством ранее. Эта информация включает в себя представления о механизмах возникновения тех или иных заболеваний, о сопровождающих эти заболевания симптомах и синдромах, об уже имеющихся лекарственных средствах для симптоматической, патогенетической или этиотропной терапии, о фармакотерапевтических и побочных реакциях организма пациента на применение определенного препарата для лечения конкретного заболевания. Использование имеющихся в распоряжении исследователей данных позволяет им выдвигать обоснованные гипотезы относительно возможных направлений поиска новых более безопасных и эффективных лекарственных средств [68]. Упомянем высказывание лауреата Нобелевской премии 1988 года по физиологии и медицине сэра Джеймса Блэка: «Самая плодотворная основа для открытия нового лекарства - начать со старого лекарства» (The most fruitful basis for the discovery of a new drug is to start with an old drug) [446].
Поскольку доступная исследователю в конкретный момент времени информацияотносительномеханизмоввозникновениятойилиинойпатологии и реакциях пациентов на применение лекарственных препаратов является неполной, выдвинутая гипотеза относительно перспективности конкретного направления разработки нового препарата может подтвердиться или не подтвердиться в ходе последующих экспериментальных доклинических и клинических исследований.
Другой Нобелевский лауреат, наш выдающийся соотечественник, Иван Петрович Павлов, в 1894 году в докладе «О неполноте современного физиологического анализа действия лекарственных веществ» на фармакологической секции Общества русских врачей так сформулировал эту мысль: «Вполне безупречная проверка в лаборатории терапевтического эмпиризма составляет, по моему мнению, весьма трудную и сейчас во многих случаях, наверное, неосуществимую задачу. В лаборатории действие средства изучается, конечно, только относительно тех функций организма, которые более или менее изучены современной физиологией. Лекарство же в руках терапевта может быть полезно как раз вследствие его отношения к таким сторонам жизненного процесса, которые еще не уловлены или не уяснены физиологией. Таким образом физиологический анализ действия лекарственных средств хронически страдает неполнотой. Поэтому одной из целей фармакологической деятельности надо считать постоянное расширение рамок
44

Глава 2. Прогнозирование свойств фармакологических веществ in silico
этого анализа, введение в постоянную систему его таких пунктов, которые все еще мало привлекают к себе внимание фармакологов» [121]. Эта мысль, сформулированная более ста лет назад, является вполне современной, если в качестве дополнения к «физиологии» рассматривать биохимию, молекулярную биологию, геномику, транскриптомику, протеомику и другие «науки XXI столетия» [13].
Таким образом, любое фармакологическое вещество, изучаемое в качестве «кандидата в лекарственные препараты», одновременно является и «реагентом» для исследования функционирования биологических систем в норме и при патологиях. К сожалению, в некоторых случаях такого рода «исследование» приводит к трагическим последствиям в клинике, как, например, упомянутая выше (в разделе 1.3) «Талидомидовая трагедия». Необходимо, однако, подчеркнуть, что компания «Хеми Грюненталь», которая вывела данный препарат на рынок в конце 50-х годов прошлого века, изучала его безопасность в соответствии с существовавшими в тот период стандартами. Однако, на мышах и крысах Талидомид не проявил тератогенного действия, которое было выявлено впоследствии при его широком медицинском применении. Следствием этого стало требование обязательного тестирования тератогенности не менее чем на трех видах экспериментальных животных (в частности, тератогенный эффект Талидомида наблюдается в эксперименте на кроликах). Детальное рассмотрение проблем с экстраполяцией на человека экспериментальных данных, полученных на животных, приведено в монографии [79].
В настоящее время поиск новых фармакологических веществ тесно связан с понятием молекулярной мишени (target). Под «мишенью» подразумевают «биологическую (макро)молекулу или надмолекулярную систему, участвующую в развитии заболевания или других (желательных или нежелательных) изменениях состояния организма, с которой связывается и взаимодействует физиологически активное вещество – лиганд. В качестве биомишеней могут выступать белки, нуклеиновые кислоты, липиды и др.» [60]. Хотя представления о молекулярных основах взаимодействия фармакологических веществ были развиты существенно позже, более ста лет назад Нобелевский лауреат Пауль Эрлих [249] сформулировал положение, согласно которому лекарственное вещество должно войти в соприкосновение с органом, клеткой или клеточной органеллой для изменения их функций: «Corpora non agunt nisi fixata».
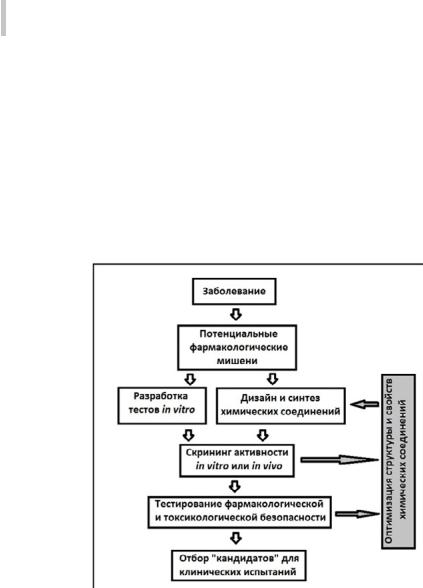
Классический процесс поиска новых фармакологических веществ можно представить схематически как совокупность экспериментальных и теоретических процедур (Рис. 5).
Вначале определяют заболевание, для которого предполагается искать потенциальные терапевтические средства, и возможные фармакологические мишени, воздействие на которые может привести к нормализации патологического процесса. Далее разрабатывают методы экспериментального тестирования биологической активности in vitro. Если связанные с заболеванием молекулярные мишени неизвестны, необходимо создать фармакологические модели для тестирования веществ на экспериментальных животных или на
45
Промышленная фармация. Путь создания продукта
изолированных органах/тканях in vivo. Осуществляется синтез соединений, сходных по структуре с веществами, для которых изучаемая биологическая активность уже была установлена, либо с их возможными аналогами (например,ингибиторыферментовсинтезируютпоаналогиисоструктуройсубстратов; агонисты и антагонисты рецепторов – по аналогии со структурой эндо- генныхлигандовит.п.).Вслучае,еслидизайнвеществ-аналоговневозможен, осуществляется случайный скрининг химических соединений, принадлежащих к различным химическим классам, что создает базу для последующей оптимизации структуры и свойств фармакологических веществ.
Приведеннаянарис.5схема,конечно,являетсячрезвычайноупрощенной. Недавно была опубликована «Динамическая карта для обучения, взаимодействия, навигации и совершенствования терапевтической разработки» [503].
Рис. 5. Классический путь поиска новых фармакологических веществ
Эта «карта» представляет в деталях мультидисциплинарный процесс поиска и разработки лекарственного препарата (Рис. 6), включающий в себя множество разноплановых задач, решаемых специалистами в различных областях науки и технологий. Из приведенной на Рис. 6 информации очевидно, что путь создания нового лекарственного препарата не является прямолинейным движением «От идеи до аптеки»: в процессе разработки приходится многократно проверять экспериментально справедливость выдвигаемых гипотез и, в случае отрицательного результата, возвращаться к предшествующим этапам работы.
46
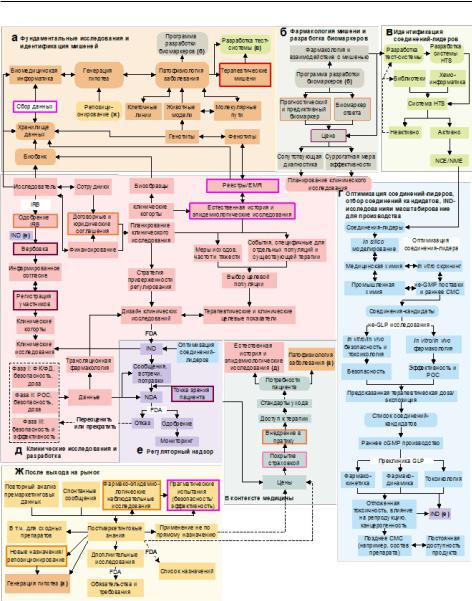
Глава 2. Прогнозирование свойств фармакологических веществ in silico
Рис. 6. Детализация современного процесса создания нового лекарственного препарата (адаптировано из работы [503]).
Список сокращений на рис. 6: CMC (Chemical Manufacturing and Control) – химическое производство и контроль (качества); EMR (Electronic Medical Record) – электронная медицинская карта; FDA (Food and Drug Administration) – надзорный орган США, контролирующий, в частности, процесс разработки лекарств и выносящий решение о допуске препаратов к применению в клинической практике; GLP (Good Laboratory Practice) – надлежащая лабораторная практика; система норм, правил и указаний,
47

Промышленная фармация. Путь создания продукта
направленных на обеспечение согласованности и достоверности резуль-
татов лабораторных исследований; GMP (Good Manufacturing Practice) –
надлежащая производственная практика; правила, которые устанавливают требования к организации производства и контроля качества лекарственных средств для медицинского и ветеринарного применения; HTS (HighThroughput Screening) – высокопроизводительный скрининг; технология, позволяющая автоматизировано проводить множество биологических экспериментов в короткие сроки; IND (Investigational New Drug) – экспериментальное новое лекарство; правовой статус лекарственного препарата, который позволяет осуществлять его транспортировку через границы США. Решение о присвоении данного статуса лекарству выносит FDA после анализа доклинических данных о его эффективности и безопасности, присвоение статуса необходимо для начала клинических исследований; IRB(InstitutionalReviewBoard)–независимыйкомитетпоэтике;NCE(New Chemical Entity) – лекарство, которое не содержит действующего вещества, одобренного FDA ранее; NDA (New Drug Application) – заявка на рассмотрение возможности применения нового лекарства в клинической практи-
ке, в США подается в FDA; NME (New Molecular Entity) – лекарство, со-
держащее (новое) действующее вещество, которое не было одобрено FDA ранее; POC (Proof Of Concept) – доказательство жизнеспособности идеи.
Тем не менее, парадигма «заболевание → фармакологическая мишень → действующий на эту мишень лиганд» является ключевой для процесса создания нового лекарственного средства. Дальнейшим развитием этих представлений являются концепция мультитаргетных препаратов (фармакологических веществ, одновременно действующих на несколько мишеней) и сетевая фармакология (рассмотрение последствий воздействия лекарственного препарата на организм с учетом особенностей регуляторных сигнальных сетей, в которые вовлечена конкретная мишень), что будет более детально представлено в последующих разделах.
Прежде всего, рассмотрим современное состояние научных знаний в области молекулярных мишеней действия лекарств.
2.2. Молекулярные мишени лекарственных препаратов
Согласно существующим оценкам, число белков человека, являющихся молекулярными мишенями разрешенных к медицинскому применению лекарств, составляет порядка 700 [456]. Однако, в геноме человека имеется около 20 тысяч генов, что с учетом альтернативного сплайсинга
ипосттрансляционных модификаций позволяет оценить число потенциальных фармакологических мишеней, как превышающее 2 млн. Помимо этого, фармакологическими мишенями являются белки бактерий, вирусов
идругих микроорганизмов. Одним из дополнительных механизмов фармакологической модуляции биологических функций является ингибиро-
48

Глава 2. Прогнозирование свойств фармакологических веществ in silico
вание белок-белковых взаимодействий [238], что, учитывая огромное число (комбинаторику) возможных попарных сочетаний белков, многократно увеличивает количество потенциальных мишеней. Кроме того, фармакологическими мишенями являются и другие биологические молекулы, в частности, ДНК и РНК, что также вносит существенный вклад в данную оценку.
Одним словом, мы имеем дело с колоссальным количеством потенциальных фармакологических мишеней, и привести более-менее обоснованные данные относительно их числа в настоящее время не представляется возможным. Можно, однако, подсчитать количества фармакологических мишеней, валидность которых для терапии конкретного заболевания подтверждена с различной степенью достоверности. Так, например, в постоянно обновляемой коммерчески доступной базе данных Clarivate Analytics Integrity [260] используются следующие категории мишеней:
V (Validated Conditions) – мишень, ассоциированная с механизмом действия одного или нескольких фармакологических веществ, находящихся в стадии активной разработки на доклинических или клинических этапах исследований или уже зарегистрированных или разрешенных к медицинскому применению для лечения рассматриваемого заболевания.
C (Candidate Conditions) – мишень, ассоциированная с механизмом действия одного или нескольких фармакологических веществ, находившаяся ранее в стадии активной разработки на доклинических или клинических этапах исследований, однако в настоящее время не разрабатываемая с целью лечения рассматриваемого заболевания.
E ( Exploratory Conditions) – мишень, ассоциированная с механизмом действия одного или нескольких фармакологических веществ, находящихся на стадии биологических испытаний с целью лечения рассматриваемого заболевания.
На 2019 г. в базе данных Integrity содержалось 2558, 4209 и 6073 мишеней, относящихся к категориям V, C и E, соответственно. 2283 мишени категории V относятся к типу «Белок» (Protein), а 275 – к типу «Ген» (Gene).3443мишеникатегории C относятсяктипу«Белок»,а766–ктипу «Ген». 4727 мишени категории E относятся к типу «Белок», а 1346 – к типу «Ген».
На рис. 7 представлены данные по числу различных категорий мишеней в2018-2019г.г.(всечислауказанына1январяконкретногогода).Приведен- ныенарисункеданныеуказываютнаинтенсивностьпроведенияисследований в этой области, что приводит к постоянному росту числа мишеней всех трех категорий. В то же время, видно, что темпы роста мишеней категории «Validated» заметно ниже по сравнению с мишенями категорий «Candidate» и «Exploratory». Это свидетельствует о том, что возникающие на ранних этапах работы гипотезы о перспективности тех или иных мишеней далеко не всегда подтверждаются в последующих доклинических и клинических исследованиях.
49

Промышленная фармация. Путь создания продукта
Рис. 7. Ростчисларазличныхкатегориймишенейв2008–2019г.г.Темнаязаливка–Validated; градиентная заливка – Candidate; штриховая заливка – Exploratory
Не все потенциальные мишени, однако, имеют одинаковые шансы быть использованными для разработки лекарственных препаратов, что послужило поводом для эмоционального заголовка статьи «Мишени, мишени повсюду – а где же новые антибактериальные препараты?» [441]. Для оценки возможности разработки фармакологических веществ, действующих на конкретную молекулярную мишень, предложен специальный термин «druggability» [229, 335], характеризующий «пригодность» мишени для поиска или конструирования действующих на него лигандов.
В Глоссарии [60] понятие «druggability» определяется следующим образом: «Пригодность биомишени для разработки лекарства – существование или возможность создания химических соединений, обладающих сродством к данной биомишени и способных произвести в результате связывания с ней благоприятный терапевтический эффект». В соответствии с данным определением,необходимоодновременноевыполнениедвухусловий.Первоеусловие связано с наличием в структуре макромолекулы-мишени сайтов, с которыми могут специфически связываться какие-либо лиганды, что позволяет разрабатыватьметодыдляпредсказания«druggability»конкретныхмишеней на основе анализа особенностей ее пространственной структуры [233, 327, 361]. Второе условие выходит за рамки только лишь наличия структурной комплементарности определенных участков макромолекулы-мишени и ее лиганда: необходимо, чтобы связавшееся с мишенью фармакологическое вещество определенным образом модулировало функцию мишени: очевидно, что эффекты ингибиторов и активаторов ферментов, агонистов и антагонистов рецепторов существенно отличаются. Более того, для проявления определенного фармакотерапевтического эффекта на уровне организма нужно, чтобы воздействие на мишень не привело к существенному изменению функционированиярегуляторнойсигнальнойсети,однимизэлементовкоторой является данная мишень [516]. Такого рода изменения возможны за счет
50