

Энергия Гиббса направленность химических процессов
Для учета одновременного влияния энтальпийного (Н) и энтропийного (S) факторов при постоянных давлении и температуре используют так называемую энергию Гиббса (свободную энергию, uзобарно-uзоmермический потенциал):
G = Н – Т S,
или для стандартных условий:
G = Н
= Н – Т
S
– Т
S , (1)
, (1)
где
G – изменение стандартной энергии Гиббса
при температуре Т.
– изменение стандартной энергии Гиббса
при температуре Т.
Расчет изменения стандартной энергии Гиббса обычно проводят по приближенному уравнению:
G
Н
Н – ТS
– ТS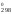 . (2)
. (2)
Обозначения:
G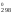 – изменение стандартной энергии Гиббса
в реакции;
– изменение стандартной энергии Гиббса
в реакции;
G – изменение стандартной энергии Гиббса
в реакции при температуре Т;
– изменение стандартной энергии Гиббса
в реакции при температуре Т;
G – стандартная энергия Гиббса образования
веществ и ионов из простых веществ. G
– стандартная энергия Гиббса образования
веществ и ионов из простых веществ. G – изменение энергии Гиббса реакции
образования 1 моля вещества из простых
веществ при условии, что все участники
реакции находятся в стандартных
состояниях.
– изменение энергии Гиббса реакции
образования 1 моля вещества из простых
веществ при условии, что все участники
реакции находятся в стандартных
состояниях.
Единицы
измерения G – кДж/моль; G
– кДж/моль; G – кДж.
– кДж.
Для
расчета G для процесса необходимо:
для процесса необходимо:
записать соответствующий процесс, указав агрегатные состояния веществ, участвующих в реакции;
расставить стехиометрические коэффициенты;
выписать из справочника величины стандартных теплот образования и стандартных энтропий всех участвующих в реакции веществ в соответствующих агрегатных состояниях;
рассчитать значения Н
 и S
и S реакций, как указано выше, и, подставив
их в уравнение G
реакций, как указано выше, и, подставив
их в уравнение G = Н
= Н – 298·S
– 298·S ,
найти значение G
,
найти значение G .
Как и любая термодинамическая функция,
энергия Гиббса является функцией
состояния, т.е. ее значение не зависит
от пути протекания процесса, а лишь от
исходного и конечного состояний системы.
Поэтому энергию
Гиббса химической реакции можно
рассчитать как сумму энергий Гиббса
образования продуктов реакции за
вычетом суммы энергий Гиббса образования
исходных веществ с учетом стехиометрических
коэффициентов.
Так как в таблицах приведены значения
G
.
Как и любая термодинамическая функция,
энергия Гиббса является функцией
состояния, т.е. ее значение не зависит
от пути протекания процесса, а лишь от
исходного и конечного состояний системы.
Поэтому энергию
Гиббса химической реакции можно
рассчитать как сумму энергий Гиббса
образования продуктов реакции за
вычетом суммы энергий Гиббса образования
исходных веществ с учетом стехиометрических
коэффициентов.
Так как в таблицах приведены значения
G ,
то использовать такой расчет можно
лишь для G
,
то использовать такой расчет можно
лишь для G .
.
При расчете G
 пренебрегают зависимостью Н0
и S0
от температуры и рассчитывают G
пренебрегают зависимостью Н0
и S0
от температуры и рассчитывают G по приближенному уравнению
по приближенному уравнению
G
Н
Н – ТS
– ТS ..(2).
..(2).
По величине изменения энергии Гиббса в реакции можно судить о принципиальной термодинамической возможности осуществления процесса.
Термодинамика утверждает, что в любой закрытой системе при постоянном давлении и температуре возможен только такой самопроизвольный процесс, который ведет к уменьшению энергии Гиббса (G <0). При значениях G >0 процесс термодинамически невозможен. При G = 0 система находится в состоянии равновенсия.
Если
условия протекания процесса отличаются
от стандартных, то используют величину
G.
Значений
G
для процесса может быть получено
множество в отличие от единственного
значения G Связь между G
и
Gо
выражается уравнением, получившим
название изотермы
Вант-Гоффа
( в честь голландского физико-химика),
которая для реакции:
Связь между G
и
Gо
выражается уравнением, получившим
название изотермы
Вант-Гоффа
( в честь голландского физико-химика),
которая для реакции:
а А + в В = с С + d D
записывается в виде:
G = Gо + RT ln (PAa PBb/ PCc PDd)
или
G = Gо + RT ln (CAa CBb/ CCc CDd)
Где Р – относительные парциальные давления соответствующих веществ; С концентрации соответствующих растворенных веществ.
Если концентрации либо парциальные давления веществ, участвующих в реакции = 1, то G = Gо.
При равновесии G = 0, а выражение под логарифмом преобразуется в Кр (Кс для растворов). Отсюда получаем одно из важнейших уравнений термодинамики, связывающее константу химического равновесия с изменением стандартной энергии Гиббса:
G = - RTlnКр,T
= - RTlnКр,T
Если в уравнение подставить значение постоянной R = 8,314 Дж/(моль К) и ввести коэффициент перехода от натурального к десятичному логарифму 2,303, то выражение можно записать следующим образом:
G = –RT
2,303lgKp
= –19,15TlgKp.
= –RT
2,303lgKp
= –19,15TlgKp.
Поскольку
G
Н
Н – ТS
– ТS ,
справедливо выражение
,
справедливо выражение
–RTlnKp
Н – ТS
– ТS .
.
Используя уравнение изотермы для любой реакции при некоторых произвольно выбранных значениях давления и температуры можно рассчитать G (бесконечное множество значений) и сделать вывод о термодинамической вероятности протекания этой реакции при выбранных условиях.
Анализ многочисленных экспериментальных данных показал, что при реальных изменениях условий (парциальных давлений или концентраций реагирующих веществ) второе слагаемое в уравнении изотермы Вант-Гоффа не превышает 40 кДж (≤ 40 кДж). Это означает, что если /GоТ/ > 40 кДж, то вывод о возможности или невозможности протекания реакции в любых реальных условиях можно сделать просто по знаку GоТ.
Процесс
термодинамически невозможен
как самопроизвольный при G >> 0 (>40
кДж).
>> 0 (>40
кДж).
Если
G << 0 (<–40
кДж),
то процесс термодинамически возможен,
протекает в прямом направлении практически
необратимо.
<< 0 (<–40
кДж),
то процесс термодинамически возможен,
протекает в прямом направлении практически
необратимо.
Значения
G от
–40 кДж до +40 кДж
соответствуют обратимым
процессам.
от
–40 кДж до +40 кДж
соответствуют обратимым
процессам.
При
условии G = 0 оба направления процессаравновероятны.
Температура,
при которой прямой и обратный процессы
равновероятны,
может быть определена в соответствии
с формулой (1):
= 0 оба направления процессаравновероятны.
Температура,
при которой прямой и обратный процессы
равновероятны,
может быть определена в соответствии
с формулой (1):
Т
=
 .
.
Вклады энтальпийного и энтропийного факторов существенно зависят от температуры.
Если Т → 0, то G
 →
Н
→
Н .
Таким образом, при низких температурах
величина и знак G
.
Таким образом, при низких температурах
величина и знак G определяются величиной и знаком Н
определяются величиной и знаком Н .
Принизких
температурах
самопроизвольно протекают, как правило,
экзотермические
реакции.
.
Принизких
температурах
самопроизвольно протекают, как правило,
экзотермические
реакции.Если Т → ∞, то G
 →
(–
Т
S
→
(–
Т
S ).
При высоких температурах величина и
знак определяются величиной и знаком
S
).
При высоких температурах величина и
знак определяются величиной и знаком
S .
При высоких
температурах
самопроизвольно протекают, как правило,
реакции, ведущие к увеличению
энтропии.
.
При высоких
температурах
самопроизвольно протекают, как правило,
реакции, ведущие к увеличению
энтропии.
Таким
образом, в зависимости от температуры
влияние либо энтальпийного, либо
энтропийного факторов на значение и
знак G и,
следовательно, на направление может
быть определяющим.
и,
следовательно, на направление может
быть определяющим.
Табл. 1. - Влияние температуры на направление химических реакций
|
Н |
S |
G |
Принципиальная возможность и условия протекания реакции |
|
-- |
++ |
- |
Возможна при любой температуре |
|
›+ |
‹- |
- |
Принципиально невозможна. Возможна в обратном направлении |
|
-- |
-- |
± |
Возможна
при низких температурах (Т
<
|
|
++ |
++ |
± |
Возможна
при высоких температурах(Т
> |
 ).
). ).
).

График зависимости G от температуры