

Cycloaddition Reactions in Organic Synthesis
.pdf58 2 Recent Advances in Palladium-catalyzed Cycloadditions involving Trimethylenemethane and its Analogs
2.2
Mechanism for [3+2] Carbocyclic Cycloaddition
The originally proposed mechanism for the methylenecyclopentane formation depicted in Scheme 2.1 involves the initial formation of a -allyl intermediate (3) which then undergoes a facile elimination of trimethylsilyl acetate to generate the palladium-bound TMM complex (2) [6]. An X-ray crystallographic analysis of a stable TMM-Pd complex prepared by a different route indicates that the ligand binds in an unsymmetrical 3 mode, in contrast to virtually all other metal complexes of this fragment which have symmetrical 4 binding mode [7]. This observation is consistent with a theoretical study that also indicates that one of the carbon termini of the TMM ligand carries a more negative charge [8]. In the methylenecyclopentane formation process, it is believed that it is this carbon terminal which Michael adds to the electron-deficient olefin to first generate an intermediate complex (4). Subsequent intramolecular cyclization of (4) gives rise to the fivemembered ring and regenerates the catalyst (Scheme 1). This stepwise, non-con- certed mechanism has withstood the test of time, as it is also used to explain the lack of stereospecificity in cycloadditions with cis olefins [1a, 9].
Scheme 2.1 Proposed mechanism for TMM [3+2] cycloaddition with an electron deficient olefin

2.3 Dynamic Behavior of TMM-Pd Complexes 59
2.3
Dynamic Behavior of TMM-Pd Complexes
A deuterium-labeling study of (1) verifies that all three methylene termini can scramble in the unsymmetrical TMM-Pd complex (2) [10]. This dynamic behavior, which can be envisioned as having the palladium center whirl around the ligand, is also manifested in substituted systems. Cycloadditions of either of the methyl precursor (5) or (6) give the same major product (9) (80–100% depending on the nature of the olefin). Again, this observation is explained by a faster equilibration (compared with cycloaddition) between the two initially formed TMM complexes (7) and (8), and that (7) is more reactive towards addition to the olefin (Scheme 2.2). Interestingly, the theoretical study by Fenske suggests that complex (7) is also favored thermodynamically [8]. The same regioselectivity is observed with other substituted systems even when the methyl group is replaced with an electron-withdrawing group, e.g. CN and CO2Me, electron-donating group, e.g. OAc, or a bulky substituent, e.g. SiMe3 [4].
Scheme 2.2 Proposed mechanism for substituted TMM [3+2] cycloaddition
Under high pressure, the reaction favors the formation of kinetic adducts (10) and (11) with precursor (6). Further regioselectivity and yield enhancement could be achieved with the bidentate phosphite ligand tpdp (12) as illustrated in Scheme 2.3 [11].

60 2 Recent Advances in Palladium-catalyzed Cycloadditions involving Trimethylenemethane and its Analogs
Scheme 2.3 Substituted TMM [3+2] cycloaddition at ambient pressure and under high pressure
2.4
Application in Organic Synthesis
2.4.1
General Comment
The use of TMM [3+2] methodology to construct five-membered carbocyclic rings offers many advantages. A wide range of electron-deficient alkenes can be used as acceptor with excellent yields. These include , -unsaturated systems such as esters, amides, ketones, nitriles, and sulfones. The cycloaddition is often highly diastereoselective and the resulting exocyclic methylene group is useful for further structural elaboration. Reaction condition simply entails heating a solution of the TMM precursor and the substrate with the palladium catalyst under reflux in an aprotic solvent under an inert atmosphere.
Recent discoveries have further expanded the synthetic potential of TMM cycloaddition approach. For example, the carbonate (13) has been developed as another convenient source of TMM. In this case, the desilylation is initiated by the alkoxide, which is generated from the decarboxylation of the carbonate leaving group (Scheme 2.4). The higher reactivity of this type of precursor can be advantageous in facilitating the cycloaddition (vide infra). Furthermore, trialkyl phosphite has been found to be more efficient than triphenylphosphine as the ligand in many cycloadditions.
Scheme 2.4 TMM-Pd formation from carbonate precursor (13)

2.4 Application in Organic Synthesis 61
Other advances include the construction of sevenand nine-membered rings via the analogous [4+3] and [6+3] cycloadditions with dienes and trienes respectively. Heterocycles, such as tetrahydrofurans and pyrrolidines, are accessible using carbonyl compounds and imines as substrates. The following discussion is organized around these recent discoveries. It serves to illustrate the versatility and the high degree of selectivity which are some of the distinctive features of the Pd-TMM chemistry.
2.4.2
[3+2] Cycloaddition: The Parent TMM
2.4.2.1 Recent Applications in Natural and Unnatural Product Synthesis
The parent TMM precursor (1), now commercially available, has played a pivotal role in the execution of many synthetic plans directed at natural and unnatural targets. Reaction of (1) with 2-(methoxycarbonyl)cyclohexenone (14, R = CO2Me) in the presence of palladium acetate and triethyl phosphite produced the adduct (15) in near quantitative yield. This cycloadduct is a critical intermediate in the total synthesis of a hydroxykempenone (16), a component of the defensive substances secreted by termites (Scheme 2.5) [12]. In accord with a previous observation by Trost that unactivated 2-cyclohexenone reacts poorly with TMM-Pd [13], the substrate (14, R = Me) was essentially inert in the cycloaddition.
Scheme 2.5 TMM [3+2] cycloaddition in the total synthesis of hydroxykempenone (16)
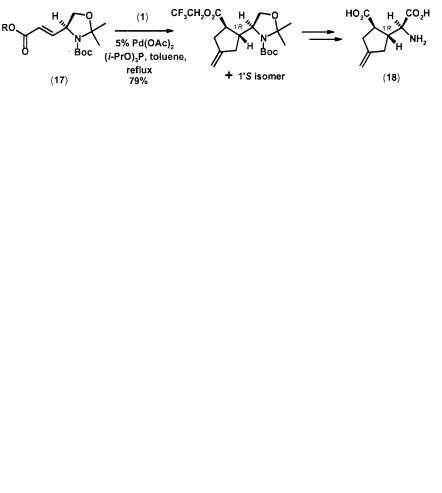
TMM cycloaddition also provides access to structurally rigid glutamate analogs. Thus (1) cycloadds to , -unsaturated ester (17, R = CH2CF3) to give a 79% yield of a mixture of 1 R and 1 S methylenecyclopentane in 8.8 : 1.2 ratio. Further purification and transformation provided the glycine (18), which was shown to be a potent agonist of kainate receptors. The more electron-deficient trifluoroethyl ester is needed for efficient cycloaddition as no reaction was observed with the corresponding methyl ester (17, R = Me) (Scheme 2.6) [14].
2.4.2.2 Novel Substrates for TMM Cycloaddition
Nitro-activated glycals and aromatic compounds are good acceptors for TMM-Pd chemistry. The nitroglycal (19) reacts with (1) to give a 2.4 : 1 ratio of products (20) and (21) in a combined yield of 69% (Eq. 2). Treatment of the less electron-rich
62 2 Recent Advances in Palladium-catalyzed Cycloadditions involving Trimethylenemethane and its Analogs
Scheme 2.6 TMM [3+2] cycloaddition in the synthesis of glutamate analog (18)
pseudoglycal (22) with the carbonate precursor (13, R = tOBu) gives a single product (23) at ambient temperature with an undisclosed yield (Eq. 3). In this case, the less reactive acetate (1) failed to participate under a variety of reaction conditions [15].
!2"
!3"
Nitroaromatics, such as 1-nitronaphthalene and 4-chloronitrobenzene, have been reported to react with the acetate (1) to produce cycloadducts arising from the subsequent elimination of HNO2 [15]. Improved yields were observed with the introduction of 1.25 equiv. of 2,6-di-t-butylpyridine as an acid scavenger, and using the corresponding pivalate of (1) as the TMM precursor. While 3-nitropyridine and phenyl triflone (PhSO2CF3) failed to participate in the cycloaddition, 5-nitroquino- line did give a good yield of adduct (24) (Scheme 2.7) [16].
Palladium-catalyzed cycloaddition of (1) to C60 has been reported to proceed in 25% yield. Interestingly, the reaction requires the C60 be first treated with (PPh3)4Pd and dppe in benzene before the introduction of (1) [17].

2.4 Application in Organic Synthesis 63
Scheme 2.7 TMM [3+2] cycloaddition with nitroaromatics
2.4.3
[3+2] Cycloaddition: Substituted TMM
2.4.3.1 Cyclopropyl-substituted TMM
Reaction of the cyclopropyl-substituted pivalate (25) with dimethyl benzylidenemalonate in the presence of a palladium catalyst gave a mixture of alkylidenecyclopropane (26) and vinylcyclopropane (27). The ratio of these two adducts is found to be quite sensitive to the choice of ligand and solvent. While triisopropyl phosphite favors the formation of the methylenecyclopropane (26), this selectivity is completely reversed with the use of the bidentate phosphite ligand dptp (12). Interestingly there was no evidence for any products that would have derived from the ring opening of the cyclopropyl-TMM intermediate (Scheme 2.8) [18].
Scheme 2.8 Cyclopropyl-TMM [3+2] cycloaddition
64 2 Recent Advances in Palladium-catalyzed Cycloadditions involving Trimethylenemethane and its Analogs
Adducts derived from cyclopropyl-TMM reactions are versatile synthetic intermediates. Alkylidenecyclopropanes have been proven useful in further Pd-cata- lyzed transformations [4]. On the other hand, vinylcyclopropanes can undergo smooth thermal ring-expansion to cyclopentenes. Thus, a total synthesis of 11-hy- droxyjasionone (27) was achieved with the cyclopropyl-TMM cycloaddition as the crucial step, and the thermal rearrangement of the initial adduct (28) as an entry to the bicyclo[6.3.0]undecyl compound (29), a key intermediate in the synthetic sequence (Scheme 2.9) [19].
Scheme 2.9 Cyclopropyl-TMM [3+2] cycloaddition in the synthesis of 11-hydroxyjasionone (27)
2.4.3.2 Phenylthio-TMM
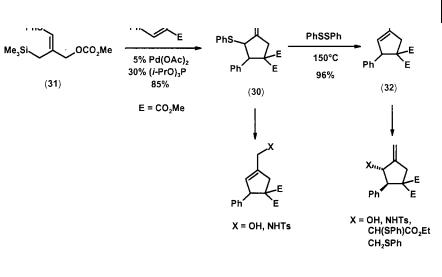
Contrary to the expectation that a sulfur-containing substituent will be a catalyst poison, a phenylthio group serves as an effective selectivity control element in TMM cycloadditions. A single regioisomer (30) was obtained from the carbonate precursor (31) in good yield. The thermodynamically more stable sulfide (32) is readily accessible from (30) via a 1,3-sulfide shift catalyzed by PhSSPh. A wide array of synthetically useful intermediates could be prepared from the sulfides (30) and (32) with simple transformations (Scheme 2.10) [20].
2.4.4
[3+2] Cycloaddition: Intramolecular Versions
2.4.4.1 Introduction and Substrate Synthesis
Intramolecular [3+2] cycloadditions, i.e., having the TMM moiety and the acceptor linked by a tether, have great synthetic utility in polycarbocycle construction. The construction of {5.5}, {6.5}, and {7.5}ring systems has been demonstrated with this methodology [21–25]. A number of efficient routes to acyclic precursors were developed (Scheme 2.11). The organometallic reagent (31), generated from 2-bro- mo-3-(trimethylsilyl)propene (32) [26], is a key component in the construction of
2.4 Application in Organic Synthesis 65
Scheme 2.10 Phenylthio-substituted TMM [3+2] cycloaddition
the TMM portion. The bifunctional aldehyde (33) proves useful in a lynchpin-type strategy to the requisite substrates.
2.4.4.2 Synthesis of Bicyclo[3.3.0]octyl Systems
Treatment of the substrate (34) with catalytic (Ph3P)4Pd and dppe provided the desired bicyclo[3.3.0]octanes (35) and the acetate elimination product (36). The choice of ligand is crucial in this case since using only dppe or Ph3P increased the amount of (36). On the other hand addition of BSA (N,O-bis(trimethylsilyl) acetamide) minimized this side product (Scheme 2.12) [24].
Intramolecular cycloadditions of substrates with a cleavable tether have also been realized. Thus esters (37a–37d) provided the structurally interesting tricyclic lactones (38–43). It is interesting to note that the cyclododecenyl system (n = 7) proceeded at room temperature whereas all others required refluxing dioxane. In each case, the stereoselectivity with respect to the tether was excellent. As expected, the cyclohexenyl (n = 1) and cycloheptenyl (n = 2) gave the syn adducts (38) and (39) almost exclusively. On the other hand, the cyclooctenyl (n = 3) and cyclododecenyl (n = 7) systems favored the anti adducts (41) and (42) instead. The formation of the endocyclic isomer (39, n = 1) in the cyclohexenyl case can be explained by the isomerization of the initial adduct (44), which can not cyclize due to ring-strain, to the other -allyl-Pd intermediate (45) which then ring-closes to (39) (Scheme 2.13) [20]. While the yields may not be spectacular, it is still remarkable that these reactions proceeded as well as they did since the substrates do contain another allylic ester moiety which is known to undergo ionization in the presence of the same palladium catalyst.

66 2 Recent Advances in Palladium-catalyzed Cycloadditions involving Trimethylenemethane and its Analogs
Scheme 2.11 Synthesis of substrates for intramolecular TMM [3+2] cycloadditions
Scheme 2.12 Intramolecular TMM [3+2] cycloaddition to bicyclo[3.3.0]octyl systems

2.4 Application in Organic Synthesis 67
2.4.4.3 Synthesis of Bicyclo[4.3.0]nonyl Systems
Perhydroindans (46) and (47) could be obtained in 73% yield from the carbonate (48) with only minor amounts of elimination product. The use of BSA and the triisopropyl phosphite-palladium acetate catalytic system provides further improvement. The low cis/trans selectivity in the formation of the first ring, and rapid subsequent cyclization account for the fact that the ratio of (46) to (47) is only 2:1 (Scheme 2.14). Even the presence of a bulky trialkylsiloxyl substituent adjacent to the vinyl sulfone moiety has only a minor influence on the cis/trans selectivity [24].
Introduction of an additional methyl group on the donor atom of TMM moiety gives a low 33% yield of the perhydroindans (49, X = H2) and (50, X = H2) with substantial production of the diene by-products [24]. However, it is still remarkable that the reaction works at all since the corresponding intermolecular cycloaddition failed. Incorporation of a carbonyl moiety adjacent to the donor carbon atom doubles the yield of the cycloadducts to 66% (Scheme 2.15). This so-called “acyl effect” works by making the donor carbon of the TMM unit “softer,” thus facilitating the initial step of the conjugate addition, as well as inhibiting base-in- duced side reactions [22].
2.4.4.4 Synthesis of Bicyclo[5.3.0]decyl Systems
The “acyl effect” proves crucial in the formation of the perhydroazulene systems: cyclization can only take place with the presence of an acyl group on the TMM portion whereas the parent hydrocarbon fails. For example, treatment of substrate (51) with the palladium catalyst gave a mixture of the bicyclic compounds (52) and (53) in 51% yield. The formation of endocyclic olefin (52) is presumed to occur when the first formed (53) was exposed to silica gel during purification [22]. This intramolecular cycloaddition strategy was utilized in a highly diastereoselective preparation of a key intermediate (54) in the total synthesis of (–)-isoclavuker- in A (55) (Scheme 2.16) [21].
2.4.5
Carboxylative Cycloadditions
The use of carbonate precursor (56) allows the introduction of a carboxylic function in the cycloadduct. The proposed mechanism involves internal delivery of a Pd-bound carbon dioxide to the TMM unit as depicted in Scheme 2.17 [27, 28].
One interesting feature of this carboxylative cycloaddition is the improved efficiency with poor acceptor such as cyclohexenone. With (1), the high basicity of the parent TMM (2) often gives rise to side reaction and therefore lower yield [29]. In the carboxylated TMM intermediate (57, Scheme 2.17), the basicity of the carbanion center is now moderated by the adjacent ester function, thus suppressing the competing basic reaction pathways and resulting in a higher yield (Scheme 2.18) [27]. This is yet another example of the “acyl effect” discussed earlier in the intramolecular cycloaddition. Another feature is the extraordinarily high degree of