

Principles and Applications of Asymmetric Synthesis
.pdf180 ALDOL AND RELATED REACTIONS
carbonyl compounds, and they are less susceptible to nucleophilic attack. Second, imine compounds tend to undergo deprotonation rather than additions. Usually, the presence of a Lewis acid is required to promote the nucleophilic attack of an allyl group on a carbonyl group. Imines, however, are susceptible to E=Z isomerization under such conditions, which complicates the stereochemical outcome of the reaction. In addition, even if the alkylation or allylation is successful, the amine thus formed will strongly bind to the Lewis acid catalyst, rendering the catalyst inactive.
To circumvent these problems, several strategies have been adopted. One is to increase the electrophilicity of the carbon atom of the CbN substrate by N- alkylation, N-oxidation, N-acylation, or N-sulfonylation to give reactive iminium salts, nitrones, acylimines, or sulfonimines. The problem associated with this method arises from the subsequent removal of the activating agent. Another method involves the activation of the CbN bond by coordination with Lewis acid or addition of an external promoter. In this method, reactive alkyl organometallic reagents can be used in imine additions. The less basic reagents such as allyl-boranes, allyl-stannanes, alkyl-coppers, or alkyl-cuprates and organocerium reagents can all be employed for this purpose.
Increasing interest is expressed in diastereoselective addition of organometallic reagents to the CbN bond of chiral imines or their derivatives, as well as chiral catalyst-facilitated enantioselective addition of nucleophiles to prochiral imines.98 The imines frequently selected for investigation include N- masked imines such as oxime ethers, sulfenimines, and N-trimethylsilylimines (150±153). A variety of chiral modi®ers, including chiral boron compounds, chiral diols, chiral hydroxy acids, N-sulfonyl amino acids, and N-sulfonyl amido alcohols 141±149, have been evaluated for their e½ciency in enantioselective allylboration reactions.68c

3.7 ASYMMETRIC ALLYLATION AND ALKYLATION OF IMINES |
181 |
Substrates:
The ®rst example of allylation of imines using dialkyl 2-allyl-1,3- dioxaborolane-4,5-dicarboxylates 110 or B-allyldiisopinocamphenyl borane 154 was reported in 1995.99 After hydrolysis of the reaction product, homoallyl primary amine was obtained in 54±90% yield and up to 73% ee (Scheme 3±52).
Scheme 3±52
Itsuno et al.68c found that among the imines 150±153, N-trimethylsilylimine derivative 153a has the highest reactivity. When it is treated with b-allylox- azaborolidine 156, a compound derived from toluenesulfonyl norephedrine 155, a product with high selectivity (92% ee) is obtained. Scheme 3±53 depicts the
Scheme 3±53
182 ALDOL AND RELATED REACTIONS
Scheme 3±54
reaction transition state. From the transition state, it seems likely that the addition of 156 proceeds with an optimized stereoelectronic interaction and minimized steric repulsion, providing the (S)-homoallylamine as the major product. In this transition state the steric repulsion between the bulky trimethyl silyl and the toluenesulfonyl group seems to be avoided.
Allylation of imines using this type of reagent has been extensively studied, and this transformation has become important for the synthesis of acyclic and cyclic amine derivatives.
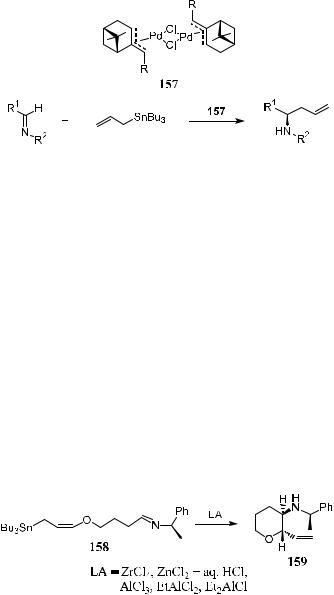
Nakamura et al.100 found that in the presence of palladium catalysts imines undergo allylation readily, providing the corresponding homoallylamines with high yields. Thus, chiral palladium complex 157 has been synthesized and applied in the asymmetric allylation of imines using allyl tributyltin as the allylation reagent. In Scheme 3±54, moderate yield and up to 82% ee have been obtained with 157 as the chiral catalyst.101
Park et al.102 demonstrated an interesting intramolecular asymmetric allylation of imine based on a substrate-controlled mode.
In the presence of ZrCl4 or HCl, cyclization of g-alkoxyallylstannane 158 bearing (R)-(‡)-1-phenylethylamine as a chiral auxiliary occurs to produce trans-b-aminocyclic ether 159 with high de (91%). As shown in Scheme 3±55, asymmetric addition of an allyl group to the imine carbon can be explained by the modi®ed Cram model 160. The attack of the allylic g-carbon approaches

3.7 ASYMMETRIC ALLYLATION AND ALKYLATION OF IMINES |
183 |
||||
|
|
|
|
|
|
|
|
|
|
|
|
Scheme 3±55
the imine carbon from a direction opposite to the Lewis acid chelating site. This orientation of the nucleophile to the Lewis acid chelating site produces the C-3 chirality of 159, giving the …2S,3R†-chirality of compound 159. The allylation of imines has been discussed elsewhere.103
Besides the allylation reactions, imines can also undergo enol silyl ether addition as with carbonyl compounds. Carbon±carbon bond formation involving the addition of resonance-stabilized nucleophiles such as enols and enolates or enol ethers to iminium salt or imine can be referred to as a Mannich reaction, and this is one of the most important classes of reactions in organic synthesis.104
Scheme 3±56 shows an example of the generation of chiral amines via nucleophilic attack onto an imine substrate in the presence of an external homochiral auxiliary. Moderate ee can be obtained from 161-induced reactions, and moderate to high ee can be expected from 162-induced reactions. For instance, when 161 (R1 ˆ Et, R2 ˆ t-Bu) is involved in the reaction, nucleophilic attack of RLi (R ˆ Me, n-Bu, and vinyl) on imine 163 gives product 164 with 81±92%
Scheme 3±56
184 ALDOL AND RELATED REACTIONS
Scheme 3±57
yield and 51±82% ee. The 162-induced reaction (R ˆ Me, n-Bu, Ph) gives product 164 with 71±91% yield and over 91% ee. Thus, in the presence of 161 or 162, various substrates derived from both aryl and alkyl aldehydes can undergo feasible organolithium nucleophilic addition reactions, and the chiral ligand can be used even in only catalytic amounts.105
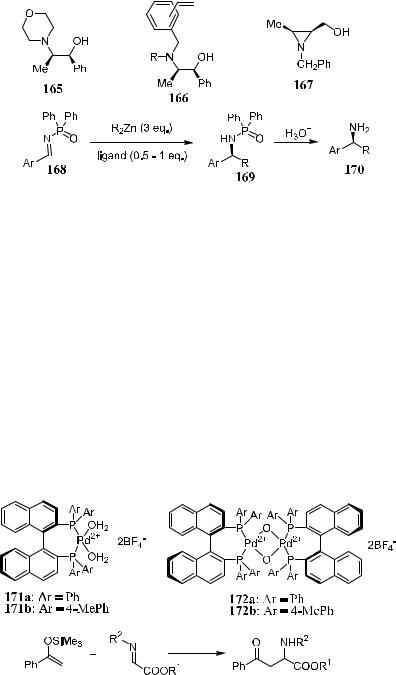
The reactivity of N-diphenylphosphinyl imines toward dialkylzinc addition in the presence of a stoichiometric or catalytic amount of chiral ligand 165, 166, or 167 has also been meticulously investigated. The reaction in Scheme 3±57 gives good yield with up to 95% ee.106
Hagiwara et al.107 reported the chiral Pd(II) complex±catalyzed asymmetric addition of enol silyl ethers to imines, based on the belief that Pd(II) enolate was involved in the reaction. They found that with compound 171a as the catalyst, very low enantioselectivity was obtained in the asymmetric reactions between silyl enol ether and imine compounds (Scheme 3±58). However, in the
Scheme 3±58

3.7 ASYMMETRIC ALLYLATION AND ALKYLATION OF IMINES |
185 |
Scheme 3±59
presence of catalyst 172, a series of enantioselective Mannich-type reactions proceeded smoothly, providing the product with up to 95% yield and up to 90% ee (Scheme 3±58, R1 ˆ i-Pr, R2 ˆ 4-MeOPh).
Ferraris et al.108 demonstrated an asymmetric Mannich-type reaction using chiral late-transition metal phosphine complexes as the catalyst. As shown in Scheme 3±59, the enantioselective addition of enol silyl ether to a-imino esters proceeds at ÿ80 C, providing the product with moderate yield but very high enantioselectivity (over 99%).
Ferraris et al.109 also studied the diastereoalkylation of a-imino esters catalyzed by Cu complexes. As depicted in Scheme 3±60, the reaction results in both high diastereoselectivity (syn/anti up to 21/1) and high enantioselectivity for the major product (ee up to 99%).
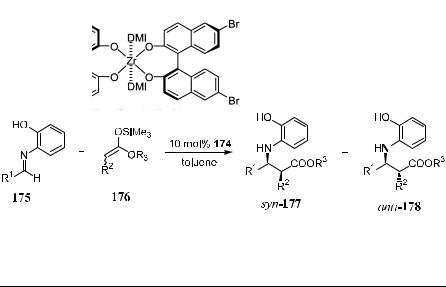
b-Amino alcohol can be found as important subunits of many bioactive compounds such as a=b-adrenergic agents or antagonists,110 HIV protease inhibitor,111 and antifungal or antibacterial peptides.112 For this reason, many attempts have been made at the asymmetric construction of b-amino alcohol subunits.113 Kobayashi et al.113 have reported a route to both synand anti-b- amino alcohols via catalytic diastereoselective or enantioselective Mannichtype reactions of a-alkoxy enolates with aldimines. By varying the protective groups on the a-alkoxy part and the ester group of the enolate part, both synand anti-selectivity can be achieved. As shown in Scheme 3±61 and Table 3±13, the expected product can be obtained in high diastereoselectivity as well as high enantioselectivity.
Scheme 3±60
186 ALDOL AND RELATED REACTIONS
Scheme 3±61
TABLE 3±13. BINOL-Zr Catalyzed Mannich-Type Reaction
|
|
|
|
Temp. |
Yield |
|
|
Entry |
R1 |
R2 |
R3 |
( C) |
(%) |
syn/anti ee (%) |
|
1 |
Ph |
(E )-TBSO |
i-Pr |
ÿ78 |
Quant. |
96:4 |
95 |
2 |
1-Nap |
(E )-TBSO |
i-Pr |
ÿ78 |
65 |
>99:1 |
91 |
3 |
2-Furyl |
(E )-TBSO |
i-Pr |
ÿ45 |
68 |
82:18 |
92 |
4 |
4-ClPh |
(E )-TBSO |
i-Pr |
ÿ78 |
73 |
92:8 |
98 |
5 |
Ph |
(Z)-BnO |
i-Pr |
ÿ45 |
Quant. |
32:68 |
95 |
6 |
Ph |
(Z)-BnO |
PMP |
ÿ45 |
91 |
6:94 |
80 |
7 |
1-Nap |
(Z)-BnO |
c-C6H11 |
ÿ45 |
80 |
8:92 |
96 |
8 |
2-Furyl |
(Z)-BnO |
PMP |
ÿ45 |
68 |
13:87 |
80 |
9 |
4-ClPh |
(Z)-BnO |
i-Pr |
ÿ45 |
Quant. |
43:57 |
91 |
10 |
4-ClPh |
(Z)-BnO |
PMP |
ÿ45 |
72 |
8:92 |
76 |
11 |
c-C6H11 |
(Z)-BnO |
c-C6H11 |
ÿ45 |
41 |
18:82 |
92 |
ee ˆ Enantiomeric excess.
Reprinted with permission by Am. Chem. Soc., Ref. 113.
3.8OTHER TYPES OF ADDITION REACTIONS: HENRY REACTION
Nitroalkane can be used as a convenient reagent for alkyl anion synthesis. Aliphatic nitro compounds can also be considered as versatile building blocks and intermediates in organic synthesis. They are readily available, and there are a wide variety of methods for converting the nitro group to other functional groups.
Acting as a strong electron-withdrawing group, the nitro group can activate a neighboring carbon±hydrogen bond for alkylation, as well as acylation or sulfonation. Through the reaction with saturated or conjugated carbonyl com-

3.8 OTHER TYPES OF ADDITION REACTIONS: HENRY REACTION |
187 |
pounds, 1,2- or 1,4-difunctionalized derivatives can be obtained, and other novel compounds can be synthesized upon oxidation, reduction, hydrolysis, and dehydration on both nitro and hydroxy groups.
Nitroalkanes can be reduced to amines, amides, hydroxylamines, nitrones, oximes, or nitriles or converted into azidosulfones and azidosul®des. More importantly, the nitro group can be converted into a carbonyl group. This allows a wide range of transformations, because it e¨ectively reverses the polarity of the neighboring carbon from nucleophilic to electrophilic. With the wide range of methods available for converting a nitro group to a carbonyl group,114 primary nitroalkanes can be considered useful and convenient intermediates in organic synthesis, particularly because they can be e½ciently used as acyl anion synthons.
The ease of converting a nitro group to a carbonyl or other functional group has signi®cantly increased the synthetic potential of nitroalkane derivatives as reagents for the nucleophilic introduction of functionalized alkyl groups in the synthesis of natural products.
The nucleophilic addition of nitroalkane to carbonyl groups is known as the Henry reaction. The products of the Henry reaction are 2-nitroalkanols,115 which are useful intermediates for nitroalkenes, 2-amino alcohols, and 2-nitro- ketones. However, this does not always give high yields because of the possible O-alkylation in preference to C-alkylation during the Henry reaction.
Scheme 3±62 is an example of an asymmetric Henry reaction reported by Sasai et al.116 in 1993. The catalyst acts in a bimetallic manner. This multifunctional e¨ect is further discussed in Chapter 8.
Scheme 3±63 gives another example in which an asymmetric Henry reaction is involved in the synthesis of the b-receptor±blocking drug (S)-propranolol.
Scheme 3±62
Scheme 3±63
188 ALDOL AND RELATED REACTIONS
Similarly, this reaction has also been applied in the synthesis of other b- receptor±blocking drugs such as (S)-metoprolol,117 (S)-pindolol,118 and the HIV protease inhibitors KNI-227 and KNI-272.119
For a review about nitroaldol and the Henry reaction, see Rosini and Ballini.120
3.9SUMMARY
This chapter has introduced the aldol and related allylation reactions of carbonyl compounds, the allylation of imine compounds, and Mannich-type reactions. Double asymmetric synthesis creates two chiral centers in one step and is regarded as one of the most e½cient synthetic strategies in organic synthesis. The aldol and related reactions discussed in this chapter are very important reactions in organic synthesis because the reaction products constitute the backbone of many important antibiotics, anticancer drugs, and other bioactive molecules. Indeed, study of the aldol reaction is still actively pursued in order to improve reaction conditions, enhance stereoselectivity, and widen the scope of applicability of this type of reaction.
3.10REFERENCES
1.For references on enantioselective aldol methodologies, see (a) Heathcock, C. H., in Trost, B. M., Fleming I. eds. Comprehensive Organic Synthesis, Pergamon Press., New York, 1991, vol. 2, Chapter 1.6, p 181. (b) Kim, B. M.; Williams, S. F.; Masamune, S., in Trost, B. M.; Fleming I. eds. Comprehensive Organic Synthesis, Pergamon Press, New York, 1991, vol. 2, Chapter 1.7, p 239. (c) Paterson, I., in Trost, B. M.; Fleming I. eds. Comprehensive Organic Synthesis, Pergamon Press, New York, 1991, vol. 2, Chapter 1.9, p 301. (d) Caine, D., in Trost, B. M.; Fleming I. eds. Comprehensive Organic Synthesis, Pergamon Press, New York, 1991, vol. 3, Chapter 1.1, p 1. (e) Braun, M.; Devant, R. Tetrahedron Lett. 1984, 25, 5031. (f ) Ambler, P. W.; Davies, S. G. Tetrahedron Lett. 1985, 26, 2129. (g) Katsuki, T.; Yamaguchi, M. Tetrahedron Lett. 1985, 26, 5807. (h) Evans, D. A.; Sjogren, E. B.; Weber, A. E.; Conn, R. E. Tetrahedron Lett. 1987, 28, 39. (i) Sasai, H.; Suzuki, T.; Itoh, N.; Shibasaki, M. Tetrahedron Lett. 1993, 34, 851. ( j) Kodota, I.; Sakaihara, T.; Yamamoto, Y. Tetrahedron Lett. 1996, 37, 3195. (k) Ito, Y.; Sawamura, M.; Hayashi, T. J. Am. Chem. Soc. 1986, 108, 6405. (l) Abiko, A.; Liu, J.; Masamune, S. J. Am. Chem. Soc. 1997, 119, 2586.
2.Eliel, E. in Morrison, J. D. ed. Asymmetric Synthesis, Academic Press, Orlando, 1983, Vol. 2, p 125.
3.Zimmerman, H. E.; Traxler, M. D. J. Am. Chem. Soc. 1957, 79, 1920.
4.Liu, J. F.; Abiko, A.; Pei, Z. H.; Buske, D. C.; Masamune, S. Tetrahedron Lett. 1998, 39, 1873.
5.Mitsui, S.; Konno, K.; Onuma, I.; Shimizu, K. J. Chem. Soc. Jpn. 1964, 85, 437.
6.Evans, D. A.; Bartroli, J.; Shih, T. L. J. Am. Chem. Soc. 1981, 103, 2127.
3.10 REFERENCES 189
7.Masamune, S.; Choy, W.; Kerdesky, F. A. J.; Imperiali, B. J. Am. Chem. Soc. 1981, 103, 1566.
8.For a new and racemization-free reductive removal of N-acyloxazolidinones, see Prashad, M.; Har, D.; Kim, H.; Repic, O. Tetrahedron Lett. 1998, 39, 7067.
9.Nerz-Stormes, M.; Thornton, E. R. Tetrahedron Lett. 1986, 27, 897.
10.(a) Heathcock, C. H. in Morrison, J. D. ed. Asymmetric Synthesis, Academic Press, New York, 1984, vol. 3, p 111. (b) Masamune, S.; Choy, W.; Petersen, J. S.; Sita,
L.R. Angew. Chem. Int. Ed. Engl. 1985, 24, 1.
11.Evans, D. A.; Bartroli, J. Tetrahedron Lett. 1982, 23, 807.
12.Evans, D. A.; Sjogren, E. B.; Bartroli, J.; Dow, R. L. Tetrahedron Lett. 1986, 27, 4957.
13.Evans, D. A.; Nelson, J. V.; Taber, T. R. in Allinger, N. L.; Eliel, E. L.; Silen, S.
H.ed. Top. Stereochem., John Wiley & Sons, New York, 1982, vol. 13, p 1.
14.Kawanami, Y.; Ito, Y.; Kitagawa, T.; Taniguchi, Y.; Katsuki, T.; Yamaguchi, M.
Tetrahedron Lett. 1984, 25, 857.
15.Ito, Y.; Katsuki, T.; Yamaguchi, M. Tetrahedron Lett. 1984, 25, 6015.
16.(a) Evans, D. A.; McGee, L. R. J. Am. Chem. Soc. 1981, 103, 2876. (b) Yamamoto, Y.; Maruyama, K. Tetrahedron Lett. 1980, 21, 4607.
17.Mukaiyama, T.; Narasaka, K.; Banno, K. Chem. Lett. 1973, 1011.
18.Gennari, C.; Bernardi, A.; Colombo, L.; Scolastico, C. J. Am. Chem. Soc. 1985, 107, 5812.
19.(a) Zelle, R. E.; DeNinno, M. P.; Selnick, H. G.; Danishefsky, S. J. J. Org. Chem. 1986, 51, 5032. (b) Danishefsky, S. J.; Selnick, H. G.; DeNinno, M. P.; Zelle, R. E.
J.Am. Chem. Soc. 1987, 109, 1572. (c) Barbier, P.; Schneider, F.; Widmer, U.
Helv. Chim. Acta 1987, 70, 1412.
20.(a) Palazzi, C.; Colombo, L.; Gennari, C. Tetrahedron Lett. 1986, 27, 1735. (b) Gennari, C.; Colombo, L.; Bertolini, G.; Schimperna, G. J. Org. Chem. 1987, 52, 2754.
21.Gennari, C.; Bernardi, A.; Scolastico, C.; Potenza, D. Tetrahedron Lett. 1985, 26, 4129.
22.Gennari, C.; Colombo, L.; Bertolini, G. J. Am. Chem. Soc. 1986, 108, 6394.
23.(a) Gennari, C.; Venturini, I.; Gislon, G.; Schimperna, G. Tetrahedron Lett. 1987, 28, 227. (b) Gennari, C.; Schimperna, G.; Venturini, I. Tetrahedron 1988, 44, 4221.
24.(a) Gennari, C.; Cozzi, P. G. J. Org. Chem. 1988, 53, 4015. (b) Gennari, C.; Cozzi,
P.G. Tetrahedron 1988, 44, 5965.
25.Poll, T.; Metter, J. O.; Helmchen, G. Angew. Chem. Int. Ed. Engl. 1985, 24, 112.
26.(a) Mikami, K.; Matsukawa, S. J. Am. Chem. Soc. 1994, 116, 4077. (b) Reetz, M. T.; Kyung, S.; Bolm, C.; Zierke, T. Chem. Ind. 1986, 824.
27.(a) Kobayashi, S.; Uchiro, H.; Shiina, I.; Mukaiyama, T. Tetrahedron 1993, 49, 1761. (b) Kobayashi, S.; Furuya, M.; Ohtsubo, A.; Mukaiyama, T. Tetrahedron Asymmetry 1991, 2, 635.
28.(a) Corey, E. J.; Cywin, C. L.; Roper, T. D. Tetrahedron Lett. 1992, 33, 6907.
(b)Parmee, E. R.; Hong, Y.; Tempkin, O.; Masamune, S. Tetrahedron Lett. 1992, 33, 1729. (c) Kiyooka, S.; Kaneko, Y.; Kume, K. Tetrahedron Lett. 1992, 33, 4927.
(d)Parmee, E. R.; Tempkin, O.; Masamune, S.; Abiko, A. J. Am. Chem. Soc.