

Principles and Applications of Asymmetric Synthesis
.pdf4.4 ENANTIOSELECTIVE DIHYDROXYLATION OF OLEFINS |
231 |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Scheme 4±33. Asymmetric dihydroxylation as a key step in the synthesis of …2S†-pro- pranolol.
Using AD mix-a or AD mix-b as the dihydroxylation agent, various ole®ns can be dihydroxylated with high ee.57,67b,72 As an example, in Scheme 4±33, aryl-allyl ethers undergo dihydroxylation yielding products with good ee. The procedure can be used as an alternative for the synthesis of …2S†-propranolol.73 Tomioka et al.74 reported an interesting example of applying chiral diamine (ÿ)-102 in the synthesis of the chromophore part of anthracycline antibiotics
(Scheme 4±34).
Scheme 4±34. Reagents and conditions: a: OsO4-(ÿ)-102/THF (96%). b: Et3SiH/ CF3COOH (78%). c: Pyridine-SO3-NEt3/DMSO (87%). d: o-C6H4(COCl)2-AlCl3/ PhNO2, (76%, 53% after recrystallization).
232 ASYMMETRIC OXIDATIONS
4.5ASYMMETRIC AMINOHYDROXYLATION
The b-amino alcohol structural unit is a key motif in many biologically important molecules. It is di½cult to imagine a more e½cient means of creating this functionality than by the direct addition of the two heteroatom substituents to an ole®n, especially if this transformation could also be in regioselective and/ or enantioselective fashion. Although the osmium-mediated75 or palladiummediated76 aminohydroxylation of alkenes has been studied for 20 years, several problems still remain to be overcome in order to develop this reaction into a catalytic asymmetric process.
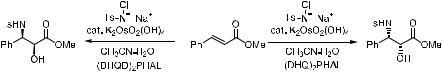
With Sharpless' recently discovered osmium-mediated asymmetric aminohydroxylation,77 this functionality can now be obtained directly from ole®ns with excellent enantioselectivities and very good yields. This process ®rst emerged as the reaction using TsNClNa (chloramine T) as the nitrogen source/ oxidant. Product a-sulfonamido hydroxy compounds can be obtained when the ole®n substrates are subjected to the aminohydroxylation reaction using chloramine-T as the nitrogen source and water as the oxygen source. In the presence of a chiral alkaloid ligand and a catalytic amount of K2OsO2(OH)4, the asymmetric aminohydroxylation reaction results in the product with good yield and enantiomeric excess. This is a process that greatly bene®ts from ligand-accelerated catalysis, as Schemes 4±35 and 4±36 illustrate.
Scheme 4±35
In contrast to the asymmetric dihydroxylation, strongly electron-de®cient alkenes are suitable substrates (Scheme 4±36). This is probably due to the greater polarizing ability of an OsbNTs group as compared with an OsbO group. Thus, acrylates in general can undergo rapid asymmetric aminohydroxylation to give the 2-hydroxy aspartic acid derivative with good results [see 103 ! 104, Scheme 4±36, (1)], although 103 is a very poor asymmetric dihydroxylation substrate. Less electron-de®cient substrates such as stilbene (107 and 109) are viable substrates, but the enantioselectivities are generally lower. (E )-alkenes, in the same situation as in the dihydroxylation process, are better substrates than (Z)-alkenes (comparing the process of 107 ! 108 with
109 ! 110).
With sulfonamide-derived chloramine salts bearing smaller organic substituents on the sulfur, for example, methanesulfonamide-derived chloramine salt 111 (chloramine-M) as the oxidant, better results are obtained. This reagent can be prepared separately and added to the reaction mixture as the stable anhydrous
|
|
|
4.5 ASYMMETRIC AMINOHYDROXYLATION |
233 |
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
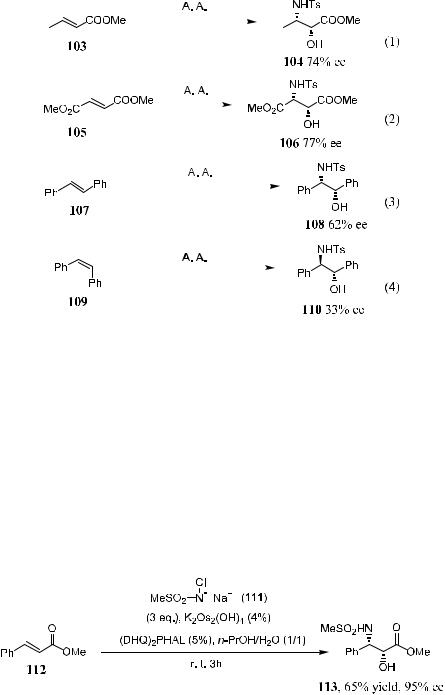
Scheme 4±36. Asymmetric aminohydroxylation.
salt, or it can be generated in situ. Thus, as illustrated in Scheme 4±37, by employing the methanesulphonamide derivative chloramine 111 as the oxidant, methyl (E)-cinnamate 112 can be converted to the corresponding a-hydroxy-b- amino product 113 with high ee (95%). Compound 113 is the taxol side chain, and this process established the shortest and the most e½cient route to the side chain of this pharmaceutically important agent.78
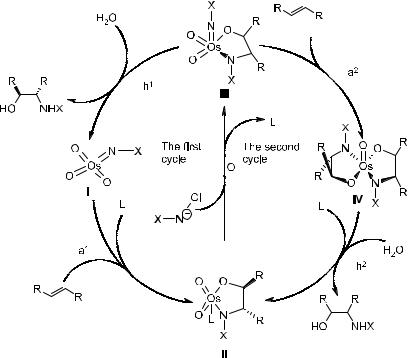
As for the mechanism of asymmetric aminohydroxylation, it has been proposed that there are at least two catalytic cycles in the reaction system (Scheme 4±38).77b It is also suggested that both electronic and steric factors play important roles in the reaction. In the ®rst cycle, in which the turnover occurs, e¨ects of the ligand on selectivity are possible. For the ligand-independent
Scheme 4±37
234 ASYMMETRIC OXIDATIONS
Scheme 4±38. Proposed mechanism for asymmetric aminohydroxylation. Sequence of steps in the ®rst catalysis cycle (left): (1) addition (a1), (2) reoxidation (O), (3) hydrolysis (h1); in the second catalysis cycle (right): (1) addition (a2), (2) hydrolysis (h2), (3) reoxidation (O). The ®rst cycle proceeds with high ee, the second with low ee. L ˆ chiral ligand; X ˆ CH3SO2ÿ.
second cycle, the ligand-mediated selectivity is lost, resulting in almost no enantioselectivity or regioselectivity. The hydrolysis step (h1) of III is expected to be easy. This makes possible the suppression of the second cycle and thus accounts for most of the rate and selectivity advantages of the methyl over the p-tosyl substituent (X).
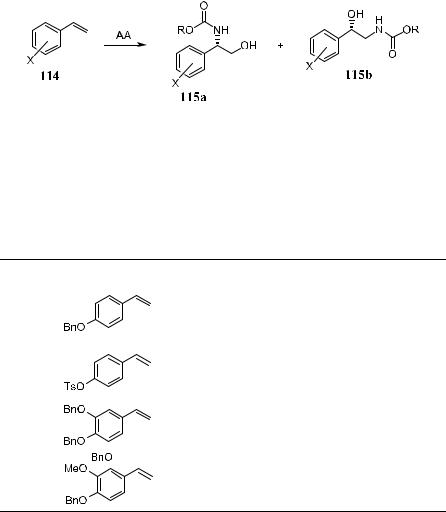
The original substrate-based asymmetric aminohydroxylation procedures, although very e½cient, display a lack of substrate scope. For example, styrenes are not suitable substrates for these reactions. Replacement of sulfonamide with alkyl carbamates (BnO2CNH2, EtO2CNH2, and t-BuO2CNH2) or amides greatly improves the reaction in the sense of both the scope of the substrate and selectivity.79 Carbamate-based nitrogen sources are especially useful because the resulting product can easily be converted to free amino alcohol. t-Butyl carbamate is superior to ethyl carbamate in terms of yield, enantioselectivity, and ease of removal of the N-protecting group. Application of the carbamate in asymmetric aminohydroxylation processes with a wide range of styrene sub-
4.5 ASYMMETRIC AMINOHYDROXYLATION |
235 |
strates79b,80 has yielded the major regioisomer in enantiomeric excess of up to 99% and yield of up to 80%. The regioselectivity is largely dependent on the substrate as well as the ligand, solvent, and ligand±solvent combination (Table 4±16).
As shown in Table 4±16, phthalazine ligand (DHQ)2DHAL in n-PrOH (alcoholic solvent) favors amine compound 115a over 115b. In CH3CN, the ratio of 115a/115b increases, and in one case the product ratio is reversed (Entry 6). The recently introduced anthraquinone (AQN) ligand appears to strongly favor this reversal of regioselectivity, especially when used in a CH3CN/H2O solvent system.
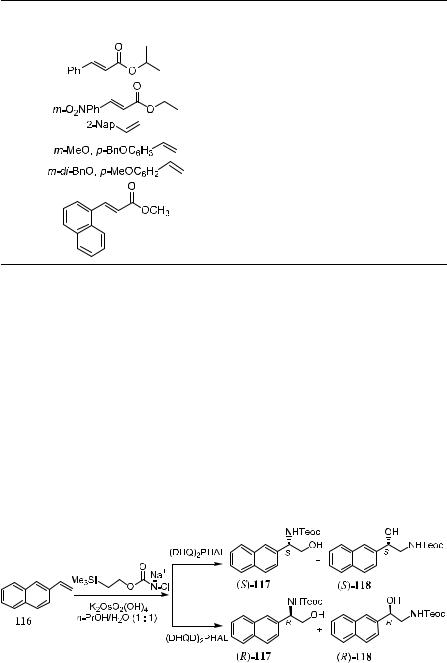
The observation that sterically less demanding nitrogen sources exhibit superior reactivity and give higher enantioselectivity has led to the new nitrogen source/oxidant 2-trimethylsilylethyl N-chloro-N-sodiocarbamate (TeoCNClNa).81 This TeoCNClNa can be prepared by reacting NaOH/
TABLE 4±16. In¯uence of Ligand and Solvent on the Regioselectivity in Asymmetric Aminohydroxylation Reaction of Four Styrene Substrates79b
Entry |
Substrate 114 |
Ligand |
Solvent |
115a:115b |
|
|
|
|
|
|
|
1 |
|
|
(DHQ)2PHAL |
n-PrOH/H2O |
88:12 |
2 |
|
|
(DHQ)2PHAL |
CH3CN/H2O |
75:25 |
3 |
|
|
(DHQ)2AQN |
n-PrOH/H2O |
33:66 |
4 |
|
|
(DHQ)2AQN |
CH3CN/H2O |
25:75 |
5 |
|
|
(DHQ)2PHAL |
n-PrOH/H2O |
50:50 |
6 |
|
|
(DHQ)2PHAL |
CH3CN/H2O |
14:86 |
7 |
|
|
(DHQ)2AQN |
n-PrOH/H2O |
17:83 |
8 |
|
|
(DHQ)2AQN |
CH3CN/H2O |
<1:50 |
9 |
|
|
(DHQ)2PHAL |
n-PrOH/H2O |
88:12 |
10 |
|
|
(DHQ)2PHAL |
CH3CN/H2O |
50:50 |
11 |
|
|
(DHQ)2AQN |
n-PrOH/H2O |
33:66 |
|
|
||||
12 |
|
|
(DHQ)2AQN |
CH3CN/H2O |
23:77 |
13 |
|
|
(DHQ)2PHAL |
n-PrOH/H2O |
77:23 |
14 |
|
|
(DHQ)2AQN |
n-PrOH/H2O |
33:66 |
15 |
|
|
(DHQ)2AQN |
CH3CN/H2O |
20:80 |
Reprinted with permission by Am. Chem. Soc., Ref. 79b.
236 ASYMMETRIC OXIDATIONS
TABLE 4±17. Asymmetric Aminohydroxylation Using TeoCNNaCl as the Nitrogen
Source
|
|
Yield for |
|
ee for (S)-117 |
ee for (R)-117 |
Entry |
Substrate |
117 (%) 117/118 |
(%) |
(%) |
|
|
|
|
|
|
|
1 |
|
70 |
>98:2 |
99 |
99 |
2 |
|
74 |
>98:2 |
99 |
98 |
3 |
|
76 |
86:14 |
97 |
96 |
4 |
|
70 |
78:82 |
99 |
99 |
5 |
|
81 |
91:9 |
98 |
97 |
6 |
|
86 |
>98:2 |
95 |
91 |
ee ˆ Enantiomeric excess.
Reprinted with permission by Pergamon Science Ltd., Ref. 81.
t-BuOCl with 2-trimethylsilyl ethyl carbamate, which can be prepared by successively adding carbonyl diimidazole and ammonia to 2-trimethylsilylethanol in benzene. TeoCNClNa substantially extends the scope of the Os-catalyzed asymmetric aminohydroxylation of alkenes. The high reactivity of this new nitrogen source/oxidant enables the reaction to proceed e¨ectively with generally better regioselectivity, enantioselectivity, and higher yields. The TeoC group can be cleaved by ¯uoride under very mild conditions, yielding the free amino alcohol with high enantiomeric purity. This result is summarized in Table 4±17.
The catalytic asymmetric aminohydroxylation of a variety of styrene derivatives, vinyl aromatics, and some other ole®ns using osmium tetroxide
Scheme 4±39
4.6 EPOXIDATION OF UNFUNCTIONALIZED OLEFINS |
237 |
in conjunction with alkaloid-derived ligands and haloamine salts has been extensively studied and used to complete the asymmetric synthesis of a- amino acids, taxol side chain, amino cyclitols, cyclohexylnorstatine, 2,3- diaminobutanoic acid, a-amino ketones, b-amino-a-hydroxy-phosphonic acid± derivatives, and diamines.82
Currently, there are seven di¨erent reagents available for carrying out asymmetric aminohydroxylations. All seven methods use a combination of osmium tetroxide [obtained from K2Os(OH)4] with alkaloid-derived ligands and the Li or Na salt of an N-halogenated sulfonamide, alkyl carbamate, or amide in an alcohol/solvent mixture. With these seven di¨erent methods in hands, chemists are in the fortunate position of being able to convert almost any ole®n into an amino alcohol with high yield and high enantioselectivity.83
4.6EPOXIDATION OF UNFUNCTIONALIZED OLEFINS
Following the success with the titanium-mediated asymmetric epoxidation reactions of allylic alcohols, work was intensi®ed to seek a similar general method that does not rely on allylic alcohols for substrate recognition. A particularly interesting challenge was the development of catalysts for enantioselective oxidation of unfunctionalized ole®ns. These alkenes cannot form conformationally restricted chelate complexes, and consequently the di¨erentiation of the enantiotropic sides of the substrate is considerably more di½cult.
4.6.1 Catalytic Enantioselective Epoxidation of Simple Ole®ns by Salen Complexes
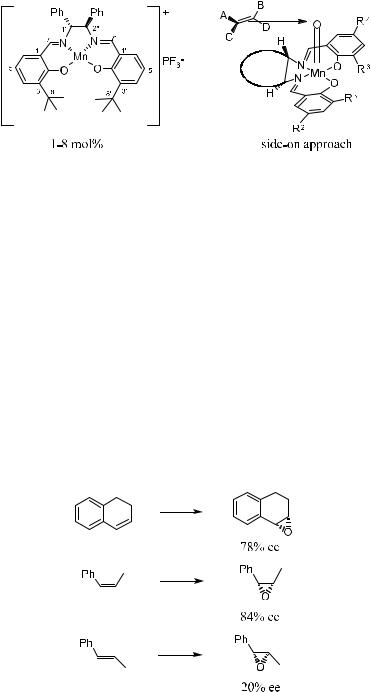
The asymmetric epoxidation of ole®ns catalyzed by chiral salen±Mn complexes was ®rst reported independently in 1990 by Zhang et al.84 and Irie et al.85 These catalysts are related to metalloporphyrin-based epoxidation catalysts, and oxygen transfer to the alkene takes place from an oxomanganese intermediate. A good example of the catalysts for asymmetric epoxidation of unfunctionalized ole®ns is the biomimetic agent related to an oxidizing enzyme, cytochrome P-450. The most commonly used oxidants are iodosylbenzene in organic solvents or sodium hypochlorite in aqueous media. Hydrogen peroxide
238 ASYMMETRIC OXIDATIONS
Figure 4±6. Side-on approach of the substrate.
in excess can also be used as an oxidant for aqueous media, but the presence of an additive ligand is essential for an e¨ective reaction. Indeed, using molecular oxygen as the oxidant is more advisable because it is the most economical and environmentally friendly oxidant.
At ®rst, the reaction was characterized as most e¨ective in the epoxidation of cis-disubstituted ole®ns.86 Later, the scope of this reaction was expanded to include the highly enantioselective synthesis of trans-disubstituted87 and trisubstituted epoxides,88 as well as certain monosubstituted epoxides.89 The ®rst example of nondirect asymmetric epoxidation of tetrasubstituted ole®ns has also appeared.90
The course of the enantioselectivity is interpreted in terms of a side-on approach by the substrate to the active oxomanganese(V) intermediate (Fig. 4±6). The results of the epoxidation are shown in Scheme 4±40.84,91 To achieve higher enantioselectivities, sterically demanding t-butyl groups substituted at the C3 and C30 positions in the salen complex have proved to be essential.
Scheme 4±40
4.6 EPOXIDATION OF UNFUNCTIONALIZED OLEFINS |
239 |
Their interactions with the more hindered side of an asymmetrical ole®n determine the orientation of the substrate during its approach to the metal±oxo bond and the subsequent enantiofacial selective oxygen transfer.
Although salen complexes of chromium, nickel, iron, ruthenium, cobalt, and manganese ions are known to serve as catalysts for epoxidation of simple ole- ®ns, the cationic Mn±salen complex is the most e½cient.
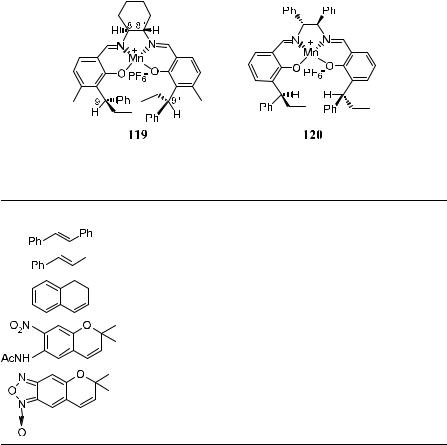
As pointed out by Hosoya et al.92 the enantiofacial selection of cis-ole®ns is mainly controlled by the asymmetric centers at the C-8(80) carbons, while that of trans-ole®ns is preferentially controlled by the asymmetric centers at the C- 9(90) carbons in 119 or 120. Optically active Mn(III)±salen complexes have catalyzed the epoxidation of cis-ole®ns with higher ee (>90%), especially when they are conjugated with an acetylene or phenyl group. However, the epoxidation of trans-ole®ns with these salen complexes shows rather poor enantioselectivity (Table 4±18).
TABLE 4±18. Epoxidation of Unfunctionalized Ole®ns Catalyzed by 119 substrate:catalyst:iodosylbenzene F1:0.025:1
Substrate |
ee (%) |
|
|
62
9 (56 when 120 is used)
91
96
94
ee ˆ Enantiomeric excess.

240 ASYMMETRIC OXIDATIONS
Jacobsen applied the salen±Mn(III) complex …R,R†-121 in the synthesis of two antihypertensive agents 122 and 12386a (Scheme 4±41) and also the taxol side chain 124.86b
Scheme 4±41. Synthesis of antihypertensive agents.
As Scheme 4±42 shows, the synthesis of 124 can be accomplished in four steps. The success of this process lies in the simplicity and low cost of all the reagents.
As an extension, this type of reagent can also be used to oxidize sul®des. Scheme 4±43 depicts the asymmetric oxidation of sul®des catalyzed by salen± Mn(III) complexes.
Iodosylarenes are impractical stoichiometric oxidants for either small-scale