

2007_-Byelorussian_Pharmacopoeia_Volume_2
.pdfКасторовое масло нерафинированное |
151 |
Серы диоксид (2.5.29). Не более 0,005 % (50 ppm).
Железо (2.4.9). Не более 0,001 % (10 ppm). 1,5 г испытуемого образца встряхивают с 15 мл
кислоты хлористоводородной разведенной Р.
Фильтруют. Фильтрат должен выдерживать испытание на железо.
Потерявмассепривысушивании(2.2.32).
Не более 20,0 %. 1,000 г испытуемого образца сушат при температуре 130°С течение 90 мин.
Сульфатная зола (2.4.14, метод А). Не бо-
лее 0,6 %. Определение проводят из 1,0 г испытуемого образца.
Микробиологическаячистота(2.6.12,2.6.13, 5.1.4). # Крахмал картофельный в условиях испытаний не обладает антимикробным действием.
Касторовое масло нерафинированное
Ricini oleum virginale
CASTOR OIL, VIRGIN
ОПРЕДЕЛЕНИЕ
Масло касторовое нерафинированное — жирное масло, полученное методом холодного прессования из семян Ricinus communis L. Может содержать подходящий антиоксидант.
ОПИСАНИЕ (свойства)
Прозрачная, почти бесцветная или светложелтая вязкая гигроскопичная жидкость.
Малорастворимо в эфире петролейном, смешивается с 96 % спиртом и кислотой уксусной ледяной.
Относительная плотность: около 0,958. Показатель преломления: около 1,479.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: D. Вторая идентификация: А, В, С.
А. Испытуемый образец выдерживает испытание «Оптическое вращение» как указано в разделе «Испытания».
В. Испытуемый образец выдерживает испытание «Гидроксильное число» как указано в разделе «Испытания».
С. Йодное число (2.5.4). От 82 до 90.
D. Испытуемый образец выдерживает испытание «Состав жирных кислот» как указано в разделе «Испытания».
ИСПЫТАНИЯ
Оптическое вращение (2.2.7). От +3,5° до
+6,0°.
Удельный показатель поглощения
(2.2.25). Не более 1,0. 1,0 г испытуемого образца растворяют в 96 % спирте Р и доводят объем
раствора этим же растворителем до 100,0 мл. Измеряют оптическую плотность полученного раствора при 269±1 нм.
Кислотное число (2.5.1). Не более 2,0.
Определение проводят из 5,0 г испытуемого образца, растворенного в 25 мл смеси указанных растворителей.
Гидроксильное число (2.5.3, метод А). Не менее 150.
Перекисное (пероксидное) число (2.5.5).
Не более 10,0.
Неомыляемые вещества (2.5.7). Не более
0,8 %. Определяют из 5,0 г испытуемого образца.
Состав жирных кислот. Газовая хромато-
графия (2.4.22) со следующими изменениями. Применяют смесь веществ для калибровки
в соответствии с таблицей 2.4.22.-3.
Испытуемый раствор. 75 мг испытуемого образца помещают в центрифужную пробирку вместимостью 10 мл с навинчивающейся крышкой и растворяют в 2 мл 1,1-диметилэтилме тило вого эфира Р1 при встряхивании и осторожном нагревании (50—60°С). В теплый раствор прибавляют 1 мл раствора 12 г/л натрия Р в метаноле безводном Р, приготовленного с необходимыми мерами предосторожности, и тщательно перемешивают не менее 5 мин. Затем прибавляют 5 мл воды дистиллированной Р,
перемешивают в течение около 30 с. Центрифугируют с ускорением 1500 g в течение 15 мин. Используют верхний слой.
Раствор сравнения. 50 мг ФСО метилрецинолеата и 50 мг ФСО метилстеарата растворяют в 10,0 мл 1,1-диметилэтилметилового эфира Р1.
Условия хроматографирования:
––колонка кварцевая капиллярная длиной 30 м и внутренним диаметром 0,25 мм, покрытая слоем макрогола 20 000 Р (толщина слоя
0,25 мкм);
––детектор: пламенно-ионизационный;
––газ-носитель: гелий для хроматографии Р;
––скорость газа-носителя: 0,9 мл/мин;
––объем вводимой пробы: 1 мкл;
––деление потока: 1:100;
––температура:
|
Время |
Температура |
|
(мин) |
(°С) |
Колонка |
0—55 |
215 |
Блок ввода проб |
|
250 |
Детектор |
|
250 |
Рассчитывают содержание каждой жирной кислоты в процентах методом внутренней нормализации.
Корректируют площадь пика, соответствующего метилрецинолеату, умножением на по-
152 |
Государственная фармакопея Республики Беларусь |
|
|
правочный коэффициент (R), рассчитанный по формуле:
R= m1 ×A2 , A1 ×m2
где:
m1 — масса навески ФСО метилрицинолеа-
та в растворе сравнения,
m2 — масса навески ФСО метилстеарата в
растворе сравнения,
A1 — площадь пика, соответствующего метилрицинолеату, вычисленная из хроматограммы раствора сравнения,
A2 — площадь пика, соответствующего метилстеарату, вычисленная из хроматограммы раствора сравнения,
Состав жирных кислот масла:
––кислота пальмитиновая: не более 2,0 %;
––кислота стеариновая: не более 2,5 %;
––кислота олеиновая и изомеры (С18:1 эквивалент длины цепи макрогола 20 000: 18,3): от
2,5 % до 6,0 %;
––кислота линолевая (С18:2 эквивалент дли-
ны цепи макрогола 20 000: 18,8): от 2,5 % до 7,0 %;
––кислота линоленовая (С18:3 эквивалент длины цепи макрогола 20 000: 19,2): не более
1,0 %;
––кислотаэйкозеновая(С20:1эквивалентдли- ны цепи макрогола 20 000: 20,2): не более 1,0 %;
––кислотарицинолеиновая(эквивалентдлины цепи макрогола 20 000: 23,9): от 85,0 % до 92,0 %;
––любая другая жирная кислота: не более
1,0 %.
Вода (2.5.12). Не более 0,3 %. Определение проводят из 5,0 г испытуемого образца.
# Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Масло касторовое в условиях испытания не обладает антимикробным действием. Перед посевом во все жидкие питательные среды и буферный раствор прибавляют 2 % по-
лисорбата 80 Р.
ХРАНЕНИЕ
В заполненном доверху воздухонепроницаемом контейнере в защищенном от света месте.
МАРКИРОВКА
Указывают наименование и концентрацию добавленного антиоксиданта.
Кокосовое масло рафинированное
Cocois oleum raffinatum
COCONUT OIL, REFINED
ОПРЕДЕЛЕНИЕ
Рафинированное кокосовое масло — жирное масло, полученное из высушенной твердой
части эндосперма Cocos nucifera L. с последующим рафинированием.
ОПИСАНИЕ (СВОЙСТВА)
Белая или почти белая пластичная масса. Практически нерастворимо в воде, легко-
растворимо в метиленхлориде и петролейном эфире (температура кипения: от 65°С до 70°С), очень мало растворимо в 96 % спирте.
Показатель преломления: около 1,449. Определение проводят при температуре 40°С.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ).
А. Испытуемый образец выдерживает испытание «Температура плавления» как указано в разделе «Испытания».
В. Испытуемый образец выдерживает испытание «Состав жирных кислот» как указано в разделе «Испытания».
ИСПЫТАНИЯ
Температура плавления (2.2.14). От 23°С
до 26°С.
Кислотное число (2.5.1). Не более 0,5.
Определение проводят из 20,0 г испытуемого образца.
Перекисное число (2.5.5). Не более 5,0. Неомыляемые вещества (2.5.7). Не более
1,0 %. Определение проводят из 5,0 г испытуемого образца.
Щелочные примеси в жирных маслах
(2.4.19). Испытуемый образец должен выдерживать испытание на щелочные примеси в жирных маслах.
Состав жирных кислот (2.4.22, метод В).
Перед испытанием кокосовое масло, осторожно нагревая, расплавляют до однородной жидкости.
Раствор сравнения. 15,0 мг ФСО трикап роина, 80,0 мг ФСО тристеарина, 0,150 г ФСО трикаприна, 0,200 г ФСО трикаприлина, 0,450 г ФСО тримиристина и 1,25 г ФСО трилаурина растворяют в смеси метиленхлорид Р — геп-
тан Р (2:8, об/об) и доводят объем раствора этой же смесью растворителей до 50 мл при нагревании при температуре от 45°С до 50°С. 2 мл полученного раствора переносят в пробирку для центрифугирования вместимостью 10 мл с закручивающейся крышкой и выпаривают растворитель в токе азота Р, растворяют в 1 мл гепта-
на Р и 1 мл диметилкарбоната Р и энергично перемешивают при постоянном нагревании (при температуре от 50°С до 60°С). К еще теплому раствору прибавляют 1 мл раствора 12 г/л на-
трия Р в метаноле безводном Р, приготовлен-
ного с необходимой осторожностью, и энергично перемешивают 5 мин. Прибавляют 3 мл воды дистиллированной Р и энергично перемешивают в течение 30 с. Центрифугируют в течение 15 мин с ускорением 1500 g. Хроматографируют 1 мкл органического слоя.
Коповидон |
153 |
Содержание жирной кислоты в процентах рассчитывают по формуле:
AХ,S,C |
×100%(м/м) , |
åAХ,S,С |
где:
Ax,s,c – скорректированная площадь пиков каждой жирной кислоты в испытуемом растворе:
AХ,S,С = AХ,S ×RC
RC – корригирующий коэффициент для пиков, соответствующих метиловым эфирам капроновой, каприловой, каприновой, лауриновой
и миристиновой кислот: |
|
|
|
R = |
mх,r ×A1,r |
, |
|
C |
A |
×m |
|
|
х,r |
1,r |
|
где:
mх,r – масса трикапроина, трикапролина, трикаприна, трилаурина или тримиристина в растворе сравнения, в миллиграммах;
m1,r – масса тристеарина в растворе сравнения, в миллиграммах;
Aх,r – площадь пиков, соответствующих метиловым эфирам капроновой, каприловой, каприновой, лауриновой и миристиновой кислот в растворе сравнения;
А1,r – площадь пика метилового эфира стеариновой кислоты в растворе сравнения;
АХ,S – площадь пиков метиловых какихлибо специфических или неспецифических эфиров жирных кислот;
RC = 1 для пиков, соответствующих каждому другому специфическому метиловому эфиру жирных кислот или какому-либо неспецифическому метиловому эфиру жирной кислоты.
Состав фракции жирных кислот должен быть следующим:
––капроновая кислота (r 0,11): не более 1,5 %;
––каприловая кислота (r 0,23): от 5,0 % до
11,0 %;
––каприновая кислота (r 0,56): от 4,0 % до
9,0 %;
––лауриновая кислота (r 0,75): от 40,0 % до
50,0 %;
––миристиновая кислота (r 0,85): от 15,0 %
до 20,0 %;
––пальмитиновая кислота (r 0,93): от 7,0 %
до 12,0 %;
––стеариновая кислота (r 1,0): от 1,5 % до
5,0 %;
––олеиновая кислота и изомеры (r 1,01): от
4,0 % до 10,0 %;
––линолевая кислота (r 1,03): от 1,0 % до 3,0 %;
––линоленовая кислота (r 1,06): не более 0,2 %;
––арахидоновая кислота (r 1,1): не более 0,2 %;
––эйкозеновая кислота (r 1,11): не более 0,2 %.
# Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Рафинированное кокосовое масло в условиях испытаний не обладает антимикробным действием.
ХРАНЕНИЕ
В заполненном доверху контейнере в защищенном от света месте.
Коповидон
Copovidonum
COPOVIDONE
H
N |
O |
H |
|
O |
O |
|
n |
|
CH3 m
n = 1,2m
(C6H9NO)n, (C4H6O2)m |
М.м. (111,1)n + (86,1)m |
ОПРЕДЕЛЕНИЕ
Коповидон представляет собой сополимер 1-этенилпирролидин-2-она и этенилацетата в массовой пропорции 3:2.
Коповидон содержит не менее 7,0 % и не более 8,0 % азота (N; А.м. 14,01) в пересчете на сухое вещество; не менее 35,3 % и не более 42,0 % этенилацетата (С4Н6О2; М.м. 86,10) в пересчете на сухое вещество.
К-значение: не менее 90,0 % и не более 110,0 % от указанного значения.
ОПИСАНИЕ (СВОЙСТВА)
Белый или желтовато-белый порошок или хлопья. Гигроскопичен.
Легкорастворим в воде, 96 % спирте и метиленхлориде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Первая идентификация: А. Вторая идентификация: В, С.
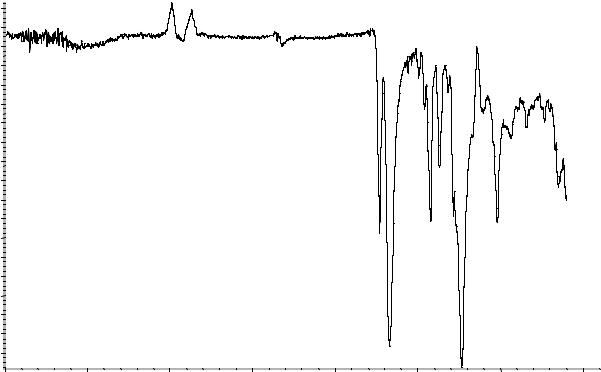
А. Инфракрасный спектр пропускания (2.2.24) испытуемого образца соответствует инфракрасному спектру ФСО коповидона # или спектру, представленному на рисунке 1.
В. К 1 мл раствора S, приготовленного как указано в разделе «Испытания», прибавляют
5 мл воды Р и 0,2 мл 0,05 М раствора йода. По-
является красное окрашивание.
С. 0,7 г гидроксиламина гидрохлорида Р
растворяют в 10 мл метанола Р, прибавляют
20 мл раствора 40 г/л натрия гидроксида Р и,
при необходимости, фильтруют. К 5 мл полученного раствора прибавляют 0,1 г испытуемого образца и кипятят в течение 2 мин. 50 мкл полученного раствора помещают на фильтроваль-
154 |
Государственная фармакопея Республики Беларусь |
|
|
П а
103,5 |
|
|
|
|
|
|
|
103,0 |
|
|
|
|
|
|
|
102,5 |
|
|
|
|
|
|
|
102,0 |
|
|
|
|
|
|
|
101,5 |
|
|
|
|
|
|
|
101,0 |
|
|
|
|
|
|
|
100,5 |
|
|
|
|
|
|
|
100,0 |
|
|
|
|
|
|
|
99,5 |
|
|
|
|
|
|
|
99,0 |
|
|
|
|
|
|
|
98,5 |
|
|
|
|
|
|
|
98,0 |
|
|
|
|
|
|
|
97,5 |
|
|
|
|
|
|
|
97,0 |
|
|
|
|
|
|
|
96,5 |
|
|
|
|
|
|
|
96,0 |
|
|
|
|
|
|
|
95,5 |
|
|
|
|
|
|
|
95,0 |
|
|
|
|
|
|
|
94,5 |
|
|
|
|
|
|
|
4000 |
3500 |
3000 |
2500 |
2000 |
1500 |
1000 |
500 |
|
|
|
В ( -1) |
|
|
|
|
Рисунок 1. Инфракрасный спектр пропускания ФСО коповидона.
ную бумагу, прибавляют 0,1 мл смеси равных объемов раствора железа (III) хлорида Р1 и кислоты хлористоводородной Р. Появляется фио-
летовое окрашивание.
ИСПЫТАНИЯ
Раствор S. 10 г испытуемого образца растворяют в воде Р и доводят объем этим же растворителем до 100 мл. Испытуемый образец прибавляют в воду малыми порциями при постоянном перемешивании.
Прозрачность (2.2.1). Раствор S по степени мутности не должен превышать эталон III.
Цветность (2.2.2, метод II). Окраска раствора S должна быть не интенсивнее эталона
В(К)5, R(Кр)5 или BY(КЖ)5.
Альдегиды. Не более 0,05 % (500 ррm)
в пересчете на ацетальдегид.
Испытуемый раствор. 1,0 г испытуемого образца растворяют в фосфатном буферном растворе рН 9,0 Р и доводят объем раствора до 100,0 мл этим же растворителем. Закрывают колбу пробкой и нагревают при температуре 60°С в течение1 ч. Охлаждают.
Раствор сравнения. 0,140 г ацетальдегидаммиака тримера тригидрата Р растворяют в воде Р и доводят объем раствора до 200,0 мл этим же растворителем. 1,0 мл полученного раствора доводят до 100,0 мл фосфатным бу-
ферным раствором рН 9,0 Р.
В 3 отдельные спектрофотометрические кюветы с толщиной слоя 1 см помещают 0,5 мл испытуемого раствора, 0,5 мл раствора сравнения
и 0,5 мл воды Р (контрольный раствор). В каждую кювету прибавляют 2,5 мл фосфатного буферного раствора с рН 9,0 Р и 0,2 мл раствора никотинамид-аденин динуклеотида Р.
Перемешивают и плотно закрывают пробкой. Выдерживают при температуре (22±2)°С в течение 2—3 мин и измеряют оптическую плотность (2.2.25) каждого раствора при 340 нм, используя воду Р в качестве компенсационного раствора. В каждую кювету прибавляют по 0,05 мл раст-
вора альдегиддегидрогеназы Р, смешивают и плотно закрывают пробкой, выдерживают при температуре (22±2)°С в течение 5 мин. Измеряют оптическую плотность каждого раствора при 340 нм, используя воду Р в качестве компенсационного раствора.
Рассчитывают содержание альдегидов по формуле:
(Аt2 -At1)-(Ab2 -Ab1)×100000×C , (As2 -As1)-(Ab2 -Ab1) m
где:
At1 —оптическаяплотностьиспытуемогораст- вора до прибавления альдегиддегидрогеназы,
Аt2 — оптическая плотность испытуемого раствора после прибавления альдегиддегидрогеназы,
Аs1 — оптическая плотность раствора сравнения до прибавления альдегиддегидрогеназы, Аs2 —оптическаяплотностьрастворасравне- ния после прибавления альдегиддегидрогеназы, Аb1 — оптическая плотность контрольного раствора до прибавления альдегиддегидрогеназы,

Коповидон |
155 |
Аb2 — оптическая плотность контрольного раствора после прибавления альдегиддегидрогеназы,
m — масса испытуемого образца в пересчете на сухое вещество, в граммах,
С — концентрация ацетальдегида в растворе сравнения, рассчитанная из массы ацетальдегидаммиака тримера тригидрата с учетом коэффициента 0,72, в мг/мл.
Пероксиды. Не более 0,04 % (400 ppm)
в пересчете на H2O2. 10 мл раствора S доводят водой Р до объема 25 мл, прибавляют 2 мл ре-
актива титана хлорида (III) и кислоты серной Р. Выдерживают 30 мин. Измеряют оптическую плотность (2.2.25) полученного раствора при 405 нм, используя в качестве раствора сравнения смесь из 25 мл раствора 40 г/л испытуемого образца и 2 мл 13 % (об/об) раствора кислоты серной Р. Оптическая плотность не должна превышать 0,35.
Гидразин. Не более 0,0001 % (1 ppm). Тонко слойнаяхроматография(2.2.27).Используютсве-
жеприготовленные растворы и ТСХ пластинки со слоем силикагеля силанизированного Р.
Испытуемый раствор. К 25 мл раствора S
прибавляют 0,5 мл раствора 50 г/л салицилово-
го альдегида Р в метаноле Р, смешивают и на-
гревают на водяной бане при 60°С в течение 15 мин. Охлаждают, прибавляют 2,0 мл ксилола Р, перемешивают в течение 2 мин и центрифугируют. Используют прозрачный слой надосадочной жидкости.
Раствор сравнения. 9 мг салицилового альдегида азина Р растворяют в ксилоле Р и дово-
дят объем раствора до 100 мл этим же растворителем. 1 мл полученного раствора разводят до
10 мл ксилолом Р.
На линию старта хроматографической пластинки наносят по 10 мкл испытуемого раствора и раствора сравнения. Помещают пластинку в хроматографическую камеру, содержащую в качестве подвижной фазы смесь вода Р — метанол Р (20:80, об/об). Когда расстояние, пройденное подвижной фазой от линии старта, составит 15 см, пластинку вынимают, высушивают на воздухе. Исследуют в ультрафиолетовом свете при 365 нм. Любое пятно, соответствующее азину салицилового альдегида, на хроматограмме испытуемого раствора должно быть не интенсивнее, чем пятно на хроматограмме раствора сравнения (не более 1 ррm).
Мономеры. Не более 0,1 %. 10,0 г испытуемого образца растворяют в 30 мл метанола Р и медленно прибавляют 20,0 мл раствора йода бромида Р. Выдерживают в защищенном от света месте в течение 30 мин, периодически перемешивая. Прибавляют 10 мл раствора 100 г/л
калия йодида Р и титруют 0,1 М раствором на-
трия тиосульфата до появления желтого окрашивания. Продолжают титрование, прибавляя
0,1 М раствор натрия тиосульфата по каплям,
до обесцвечивания раствора. Параллельно проводят контрольный опыт.
Натитрованиедолжнопойтинеболее1,8мл
0,1 М раствора натрия тиосульфата.
Примесь А. Не более 0,5 %. Жидкостная хроматография (2.2.29).
Испытуемый раствор. 100 мг испытуемого образца растворяют в воде Р и доводят объем раствора этим же растворителем до 50,0 мл.
Раствор сравнения. 100 мг 2-пирролидона Р растворяют в воде Р и доводят объем раствора этим же растворителем до 100 мл. 1,0 мл полученного раствора доводят водой Р до объ-
ема 100,0 мл.
Условия хроматографирования:
––предколонка длиной 0,025 м и внутренним диаметром 4 мм, заполненная силикагелем октадецилсилильным эндкепированным для хроматографии Р с размером частиц 5 мкм;
––колонка из нержавеющей стали длиной 0,25 м и внутренним диаметром 4,0 мм, запол-
ненная сферическим силикагелем аминогексадецилсилильным для хроматографии Р с раз-
мером частиц 5 мкм;
––температура: 30°С;
––подвижная фаза: вода Р, доведенная до рН 2,4 кислотой фосфорной Р;
––скорость подвижной фазы: 1 мл/мин;
––спектрофотометрический детектор,
длина волны 205 нм. Детектор устанавливают между предколонкой и аналитической колонкой. Второй детектор устанавливают после аналитической колонки;
––объем вводимой пробы: 10 мкл. После вы-
хода примеси А из предколонки (примерно 1,2 мин) переключают поток напрямую от насоса к аналитической колонке. До начала следующей хроматограммы промывают предколонку противотоком.
Площадь пика примеси А на хроматограмме испытуемого раствора не должна превышать площадь соответствующего пика на хроматограмме раствора сравнения (0,5 %).
Тяжелые металлы (2.4.8, метод А). Не бо-
лее 0,002 % (20 ppm). 12 мл раствора S должны выдерживать испытание на тяжелые металлы. Эталон готовят с использованием эталонного раствора свинца (2 ppm Pb) Р.
Потерявмассепривысушивании(2.2.32).
Не более 5,0 %. 0,500 г испытуемого образца сушат при температуре от 100°С до 105°С.
Сульфатная зола (2.4.14, метод А). Не бо-
лее 0,1 %. Определение проводят из 1,0 г испытуемого образца.
Вязкость, обозначенная как К-значение.
5,0 мл раствора S доводят водой Р до объема 50,0 мл. Выдерживают 1 ч и определяют вязкость (2.2.9) раствора при 25°С, используя вис

156 |
Государственная фармакопея Республики Беларусь |
|
|
козометр № 1 с минимальным временем вытекания 100 с.
Рассчитывают К-значение по формуле:
1,5×logh-1 |
+ |
|
300×c×logh+(c+1,5×c×logh |
)2 |
, |
0,15+0,003×c |
|
0,15×c+0,003×c2 |
|||
|
|
|
|||
где:
с — концентрация испытуемого образца в пересчете на безводное вещество, в г/100 мл;
h — вязкость раствора относительно вязко-
сти воды Р.
#Остаточные количества органических растворителей (2.4.24). Испытуемый образец должен выдерживать требования статьи (5.4).
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Коповидон в условиях испытания не обладает антимикробным действием.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Этенилацетат. Определяют число омыления (2.5.6) из 2,00 г испытуемого образца. Рассчитывают содержание этенилацетата в процентах, умножая полученный результат на 0,1534.
Азот. Проводят определение азота (2.5.9), используя 30,0 мг испытуемого образца и 1 г смеси из 3 частей меди сульфата Р и 997 частей калия сульфата Р. Нагревают до образования прозрачного светло-зеленого раствора и продолжают нагревать в течение 45 мин.
ХРАНЕНИЕ В воздухонепроницаемом контейнере.
МАРКИРОВКА Указывают К-значение.
ПРИМЕСИ
H
N O
А. Пирролидин-2-он (2-пирролидон).
# Красный очаровательный
E-129
ALLURA RED AC, E-129
|
|
|
|
|
|
|
H3CO |
|||||||
|
|
|
N |
|
|
N |
|
|
|
|
|
|
|
|
|
|
|
|
|
OH |
|||||||||
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
CH2SO3Na |
||||||
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NaO3S |
|
|
|
|
|
|
|
|
|
|
||||
C18H14N2Na2O8S2 |
|
|
|
|
|
М.м. 496,4 |
||||||||
ОПРЕДЕЛЕНИЕ
Краситель красный очаровательный содержит не менее 85,0 % динатрия 2-гидрокси- 1-(2-метокси-5-метил-сульфонатофенилазо)- нафтален-6-сульфоната в пересчете на сухое вещество.
ОПИСАНИЕ (СВОЙСТВА)
Мелкодисперсный порошок темно-фиоле тового цвета, без запаха.
Хорошо растворим в воде.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Спектр поглощения (2.2.25) испытуемого раствора, приготовленного как указано в испытании «Количественное определение», в области от 350 нм до 650 нм имеет максимум при
500±3 нм.
ИСПЫТАНИЯ
Раствор S. 5,0000 г испытуемого образца помещают в фарфоровую чашку, добавляют около 0,43 г магния оксида Р и 8,6 мл спиртового раствора 50 г/л магния нитрата Р. Полученную смесь тщательно перемешивают до равномерного распределения компонентов.
Чашку помещают на водяную баню при температуре от 80°С до 100°С и выпаривают досуха, после чего переносят на электроплитку и обугливают при слабом нагреве до прекращения выделения дыма. Затем чашку помещают в муфельную печь при температуре 250°С, повышают температуру до 450°С со скоростью 50°С/ч и продолжают сжигать при этих условиях до получения серой золы (приблизительно
10—15 ч).
Чашку с золой вынимают из муфельной печи, охлаждают до комнатной температуры и смачивают содержимое по каплям минималь-
ным количеством воды для хроматографии Р. Воду выпаривают досуха на водяной бане с последующей выдержкой в сушильном шкафу при температуре 140°С или на электроплитке со слабым нагревом. После охлаждения чашку с навеской снова помещают в охлажденную муфельную печь. Постепенно доводят температуру до 300°С и выдерживают в течение 30 мин. Указанную процедуру повторяют несколько раз до получения золы белого или слегка окрашенного цвета, без обугленных частиц.
В тигель с золой прибавляют 5—6 мл кис-
лоты азотной, свободной от свинца и кадмия,
Р, накрывают часовым стеклом и нагревают на электроплитке со слабым нагревом до растворения золы. Полученный минерализат количественнопереносятспомощьюсоответствующего растворителя, используемого для приготовления эталонных растворов, в мерную колбу вместимостью 10 мл, предварительно фильтруя через мембранный фильтр.

# Красный очаровательный |
157 |
Мышьяк (2.2.23, метод 2). Не более
0,0003 % (3 ppm). Раствор S должен выдерживать испытание на мышьяк.
Источник излучения: лампа с полым като-
дом для определения мышьяка. Коррекция фона: корректор Зеемана. Атомизатор: графитовая печь.
Длина волны: 193,7 нм.
Эталонные растворы готовят разбавлением ФСО или ГСО раствора ионов мышьяка 0,125
М раствором кислоты азотной, свободной от свинца и кадмия, Р (125 мл 1 М раствора кислоты азотной, свободной от свинца и кадмия, Р доводят водой для хроматографии Р до объема
1000,0 мл).
Хром (2.2.23, метод 2). Не более 0,01 % (100 ppm). Раствор S должен выдерживать испытание на хром.
Источник излучения: лампа с полым като-
дом для определения хрома.
Коррекция фона: дейтериевая лампа. Атомизатор: воздушно-ацетиленовое пламя.
Длина волны: 357,9 нм.
Эталонные растворы готовят разбавлением ФСО или ГСО раствора ионов хрома 0,125 М
раствором кислоты хлористоводородной Р (125,0 мл 1 М раствора кислоты хлористоводо-
родной доводят водой Р до объема 1000,0 мл).
Свинец (2.2.23, метод 2). Не более 0,001 % (10 ppm). Раствор S должен выдерживать испытание на свинец.
Источник излучения: лампа с полым като-
дом для определения свинца. Коррекция фона: дейтериевая лампа.
Атомизатор: воздушно-ацетиленовое пламя.
Длина волны: 217,0 нм.
Эталонные растворы готовят разбавлением ФСО или ГСО раствора ионов свинца 0,125 М
раствором кислоты хлористоводородной Р (125,0 мл 1 М раствора кислоты хлористоводо-
родной доводят водой Р до объема 1000,0 мл).
Ртуть (2.2.23, метод 2). Не более 0,0001 % (1 ppm). Раствор S должен выдерживать испытание на ртуть.
Источник излучения: лампа с полым като-
дом для определения ртути.
Коррекция фона: дейтериевая лампа. Атомизатор: метод холодного пара.
Длина волны: 253,7 нм.
Эталонные растворы готовят разбавлением ФСО или ГСО раствора ионов ртути 5 % (об/об)
раствором кислоты хлористоводородной, свободной от свинца, Р.
Кадмий(2.2.23,метод2).Неболее0,0001 % (1 ppm). Раствор S должен выдерживать испытание на кадмий.
Источник излучения: лампа с полым като-
дом для определения кадмия. Коррекция фона: корректор Зеемана. Атомизатор: графитовая печь.
Длина волны: 228,8 нм.
Эталонные растворы готовят разбавлением ФСОилиГСОраствораионовкадмия0,125Мрас-
твором кислоты азотной, свободной от свинца и кадмия,Р(125мл1Мрастворакислотыазотной, свободной от свинца и кадмия, Р доводят водой для хроматографии Р до объема 1000,0 мл).
Тяжелые металлы (2.4.8, метод А). Не бо-
лее 0,004 % (40 ppm). 2 мл раствора S доводят водой Р до 20 мл. Полученный раствор должен выдерживать испытание на тяжелые металлы. Эталон готовят с использованием эталонного раствора свинца (2 ррm Pb) Р.
Потерявмассепривысушивании(2.2.32).
Не более 5,0 %. 1,000 г испытуемого образца сушат при температуре от 100°C до 105°С.
Нерастворимые в воде вещества. Не бо-
лее 0,2 %. 5,0 г испытуемого образца растворяют
в100 мл воды Р. Фильтруют через предварительно высушенный и взвешенный стеклянный фильтр (ПОР 40). Остаток на фильтре промывают тремя порциями воды Р по 10 мл. Масса осадка, полученного после промывания и высушивания фильтра при температуре от 100°C до 105°С, не должна превышать 10 мг.
Вещества, извлекаемые эфиром. Не бо-
лее 0,2 %. 2,000 г испытуемого образца высушивают при температуре от 100°C до 105°С до постоянной массы, взбалтывают с 200 мл эфира Р
втечение 30 мин, фильтруют через беззольный фильтр в мерную колбу емкостью 200 мл, доводят объем эфиром Р до метки и перемешивают. 100 мл фильтрата переносят в высушенный до постоянной массы и взвешенный бюкс. Эфир отгоняют и остаток высушивают при температуре от 100°C до 105°С до постоянной массы. Масса остатка не должна превышать 2 мг.
Остаточные количества органических растворителей (2.4.24). Испытуемый образец должен выдерживать требования статьи (5.4).
Микробиологическаячистота(2.6.12,2.6.13, 5.1.4). Красный очаровательный в условиях испытаний не обладает антимикробным действием.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
0,100 г испытуемого образца растворяют в
воде дистиллированной Р и доводят объем раст-
вора этим же растворителем до 100,0 мл. Фильтруют через стеклянный фильтр (ПОР 40), отбрасывая первые 10—15 мл фильтрата (если образец содержит нерастворимые компоненты, раствор центрифугируют перед разведением). К 1,0 мл полученного раствора прибавляют 2 мл раствора 196,6 г/л аммония ацетата Р и доводят объем раствора водой дистиллированной Р до 100,0 мл.
Измеряют оптическую плотность (2.2.25) полученного раствора при 500 нм в кювете с толщиной слоя 10 мм, используя в качестве компенсационного раствора смесь из воды дис-
тиллированной Р и раствора 196,6 г/л аммония ацетата Р (1:1).

158 |
Государственная фармакопея Республики Беларусь |
Содержание C18H14N2Na2O8S2 в процентах рассчитывают по формуле:
A×10× f ×100 , m×540
где:
А — оптическая плотность испытуемого раствора при 500 нм;
540 — удельный показатель поглощения
C18H14N2Na2O8S2 при 500 нм; f — фактор разведения;
m — масса навески испытуемого образца, г.
Кремния диоксид коллоидный безводный
Silica colloidalis anhydrica |
|
SILICA, COLLOIDAL ANHYDROUS |
|
SiO2 |
М.м. 60,1 |
ОПРЕДЕЛЕНИЕ
Кремния диоксид коллоидный безводный содержит не менее 99,0 % и не более 100,5 % SiO2 в пересчете на прокаленное вещество.
ОПИСАНИЕ (СВОЙСТВА)
Легкий тонкий аморфный порошок белого цвета с размером частиц около 15 нм.
Практически нерастворим в воде и минеральных кислотах, кроме фтористоводородной кислоты. Растворяется в горячих растворах гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
Около 20 мг испытуемого образца дают реакцию на силикаты (2.3.1).
ИСПЫТАНИЯ
pH (2.2.3). От 3,5 до 5,5. 1,0 г испытуемого образца встряхивают с 30 мл воды, свободной от углерода диоксида, Р. Измеряют рН получен-
ной суспензии.
Хлориды(2.4.4).Неболее0,025 %(250ppm).
К1,0гиспытуемогообразцаприбавляютсмесьиз
20 мл азотной кислоты разведенной Р и 30 мл
воды Р, нагревают на водяной бане в течение 15 мин, часто встряхивая. При необходимости доводят водой Р до объема 50 мл, фильтруют и охлаждают. 10 мл фильтрата доводят водой Р до объема 15 мл. Полученный раствор должен выдерживать испытание на хлориды.
Тяжелые металлы (2.4.8, метод А). Не более 0,0025 % (25 ppm). 2,5 г испытуемого об-
разца суспендируют в достаточном количестве воды Р для получения полужидкой смеси. Высушивают при температуре 140°С. Когда сухое вещество становится белым, массу измельчают стеклянной палочкой. Прибавляют 25 мл
1 М раствора кислоты хлористоводородной
и осторожно кипятят в течение 5 мин, часто перемешивая суспензию стеклянной палочкой. Центрифугируют в течение 20 мин и фильтруют надосадочную жидкость через мембранный фильтр. К остатку в пробирке для центрифугирования прибавляют 3 мл хлористоводородной кислоты разведенной Р, 9 мл воды Р и кипятят.
Центрифугируют в течение 20 мин и фильтруют надосадочную жидкость через тот же мембранный фильтр. Остаток промывают небольшими порциями воды Р, объединяют фильтраты и промывные воды и доводят водой Р до объема 50 мл. К 20 мл раствора прибавляют 50 мг аскор-
биновой кислоты Р и 1 мл раствора аммиака концентрированного Р. Нейтрализуют раствором аммиака разведенным Р2. Доводят объем раствора до 25 мл водой Р. 12 мл полученного раствора должны выдерживать испытание на тяжелые металлы. Эталон готовят с использовани-
ем эталонного раствора свинца (1 ppm Pb) Р.
Потерявмассеприпрокаливании.Неболее
5,0 %. 0,200 г испытуемого образца прокаливают в платиновомтиглепритемпературе900°Свтечение 2 ч. Перед взвешиванием охлаждают в эксикаторе.
#Остаточные количества органических растворителей (2.4.24). Испытуемый образец должен выдерживать требования статьи (5.4).
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Кремния диоксид коллоидный безводный в условиях испытания не обладает антимикробным действием.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
Костатку,полученномувиспытании«Потеря
вмассе при прокаливании», прибавляют 0,2 мл
кислоты серной Р и 96 % спирт Р в количестве,
достаточном для полного смачивания остатка.
Прибавляют 6 мл кислоты фтористоводород-
ной Р и выпаривают досуха на плитке при температуре от 95°С до 105°С, не допуская разбрызгивания. Стенки чашки промывают 6 мл кислоты фтористоводородной Р и выпаривают досуха.
Прокаливают при температуре 900°С, охлаждают в эксикаторе и взвешивают.
Количество SiO2 в навеске испытуемого образца рассчитывают как разность между массой остатка, полученного при проведении испытания «Потеря в массе при прокаливании», и массой окончательного остатка.
Кремния диоксид коллоидный гидратированный
Silica colloidalis hydrica
SILICA, COLLOIDAL HYDRATED
ОПРЕДЕЛЕНИЕ
Кремния диоксид коллоидный гидратированный содержит не менее 98,0 % и не более

Кроскармеллоза натрия |
159 |
100,5 % SiO2 (М.м. 60,1) в пересчете на прока-
ленное вещество.
ОПИСАНИЕ (СВОЙСТВА)
Легкий мелкий аморфный порошок белого или почти белого цвета.
Практически нерастворим в воде и минеральных кислотах, кроме фтористоводородной кислоты. Растворяется в горячих растворах гидроксидов щелочных металлов.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. Около 20 мг испытуемого образца дают реакцию на силикаты (2.3.1).
В. При нагревании в сушильном шкафу при температуре от 100°С до 105°С в течение 2 ч потеря в массе составляет более 3 %.
ИСПЫТАНИЯ
Раствор S. К 2,5 г испытуемого образца при-
бавляют 50 мл кислоты хлористоводородной Р
иперемешивают. Нагревают на водяной бане в течение 30 мин, периодически перемешивая. Доводят до первоначального объема кислотой хлористоводородной разведенной Р. Выпарива-
ют досуха. К остатку прибавляют смесь из 8 мл
кислоты хлористоводородной разведенной Р и
24 мл воды Р. Нагревают до кипения и фильтруют при пониженном давлении через стеклянный фильтр (ПОР 16). Остаток на фильтре промывают горячей смесью из 3 мл кислоты хлори-
стоводородной разведенной Р и 9 мл воды Р.
Промывают небольшими порциями воды Р, объединяют фильтрат и промывные воды и доводят водой Р до объема 50 мл.
pH (2.2.3). От 4,0 до 7,0. 1,0 г испытуемого образца суспендируют в 30 мл раствора 75 г/л калия хлорида Р. Измеряют рН полученной суспензии.
Водопоглощающая способность. 5 г ис-
пытуемого образца растирают в ступке, прибавляя по каплям 5 мл воды Р. Смесь должна оставаться порошкообразной.
Вещества, растворимые в хлористоводородной кислоте. Не более 2,0 %. 10,0 мл раст-
вора S помещают в платиновую чашку, выпаривают досуха и высушивают при температуре от 100°С до 105°С до постоянной массы. Масса остатка не должна превышать 10 мг.
Хлориды (2.4.4). Не более 0,1 %. 0,5 г ис-
пытуемого образца смешивают с 50 мл воды Р
инагревают на водяной бане в течение 15 мин. Доводят водой Р до объема 100 мл и центрифугируют с ускорением 1500 g в течение 5 мин. 10 мл надосадочной жидкости доводят водой Р до объема 15 мл. Полученный раствор должен выдерживать испытание на хлориды.
Сульфаты (2.4.13). Не более 1 %. 2 мл раствора S доводят водой дистиллированной Р
до объема 100 мл. 15 мл полученного раствора должны выдерживать испытание на сульфаты.
Железо (2.4.9). Не более 0,03 % (300 ppm). К 2 мл раствора S прибавляют 28 мл воды Р. 10 мл полученного раствора должны выдерживать испытание на железо.
Тяжелые металлы (2.4.8, метод А). Не бо-
лее 0,0025 % (25 ppm). К 20 мл раствора S при-
бавляют 50 мг гидроксиламина гидрохлорида Р и 1 мл раствора аммиака концентрированно-
го Р. Доводят значение рН до 3,5 (контролируют потенциометрически) раствором аммиака раз-
веденным Р2. Доводят водой Р до объема 25 мл. 12 мл полученного раствора должны выдерживать испытание на тяжелые металлы. Эталон готовят с использованием эталонного раствора свинца (1 ppm Pb) Р.
Потеря в массе при прокаливании. Не более 20,0 %. 0,200 г испытуемого образца нагревают в платиновом тигле при температуре от 100°С до 105°С в течение 1 ч и затем прокаливают при 900°С в течение 2 ч.
#Остаточные количества органических растворителей (2.4.24). Испытуемый образец должен выдерживать требования статьи (5.4).
#Микробиологическая чистота (2.6.12, 2.6.13, 5.1.4). Кремния диоксид коллоидный гидратированный в условиях испытания не обладает антимикробным действием.
КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ
К остатку, полученному в испытании «Потеря в массе при прокаливании», прибавляют 0,2 мл кислоты серной Р и 96 % спирт Р в количестве, достаточном для полного смачивания остатка. Прибавляют 6 мл кислоты фтористоводородной Р и выпаривают досуха на плитке при температуре от 95°С до 105°С, не допуская разбрызгивания. Стенки чашки промывают 6 мл кислоты фтористоводородной Р и выпаривают досуха. Прокаливают при температуре 900°С, охлаждают в эксикаторе и взвешивают.
Количество SiO2 в навеске испытуемого образца рассчитывают как разность между массой остатка, полученного при проведении испытания «Потеря в массе при прокаливании», и массой окончательного остатка.
Кроскармеллоза натрия
Carmellosum natricum conexum
CROSCARMELLOSE SODIUM
ОПРЕДЕЛЕНИЕ |
|
Кроскармеллоза натрия — это |
натрие- |
вая соль перекрестно-сшитой, |
частично |
О-карбоксиметилированной целлюлозы.

160 |
Государственная фармакопея Республики Беларусь |
|
|
ОПИСАНИЕ (СВОЙСТВА)
Порошок белого или серовато-белого цвета. Практически нерастворима в ацетоне, эта-
ноле и толуоле.
ПОДЛИННОСТЬ (ИДЕНТИФИКАЦИЯ)
А. 1 г испытуемого образца встряхивают с
100 мл раствора 0,004 г/л (4 ppm) метиленового синего Р и дают отстояться. Испытуемый образец абсорбирует метиленовый синий и оседает в виде синей волокнистой массы.
В. 1 г испытуемого образца встряхивают с 50 мл воды Р. Переносят 1 мл полученной смеси в пробирку, прибавляют 1 мл воды Р и 0,05 мл свежеприготовленного раствора 40 г/л
α-нафтола Р в метаноле Р. Наклоняют пробир-
ку и осторожно прибавляют 2 мл кислоты серной Р вниз по стенке так, чтобы она образовала нижний слой. На границе раздела появляется красновато-фиолетовое окрашивание.
С. Испытуемый образец дает реакцию (а) на натрий (2.3.1).
ИСПЫТАНИЯ
рН (2.2.3). От 5,0 до 7,0. 1 г испытуемого образца встряхивают с 100 мл воды, свободной от углерода диоксида, Р в течение 5 мин. Измеря-
ют рН полученной суспензии.
Натрия хлорид и натрия гликолят. Сум-
марное процентное содержание натрия хлорида
инатрия гликолята не должно превышать 0,5 %
впересчете на сухое вещество.
Натрия хлорид. 5,00 г испытуемого образца помещают в коническую колбу вместимостью 250 мл, прибавляют 50 мл воды Р и 5 мл раст-
вора водорода пероксида концентрированно-
го Р и нагревают на водяной бане в течение 20 мин, периодически перемешивая для достижения полной гидратации. Охлаждают, прибав-
ляют 100 мл воды Р и 10 мл кислоты азотной Р.
Титруют при постоянном перемешивании 0,05 М
раствором серебра нитрата потенциометри-
чески (2.2.20). Используют серебряный индикаторный электрод и двойной электрод сравнения, содержащий раствор 100 г/л калия нитрата Р во внешней ячейке и стандартный раствор во внутренней ячейке.
1 мл 0,05 М раствора серебра нитрата со-
ответствует 2,922 мг NaCl.
Натрия гликолят. 0,500 г испытуемого об-
разца в пересчете на сухое вещество помещают
влабораторный стакан вместимостью 100 мл,
прибавляют 5 мл кислоты уксусной ледяной Р
и5 мл воды Р и перемешивают до полной гид ратации (около 15 мин). Прибавляют 50 мл аце-
тона Р и 1 г натрия хлорида Р. Перемешивают
втечение нескольких минут до полного осаждения карбоксиметилцеллюлозы. Фильтруют через быстрофильтрующую фильтровальную бумагу, пропитанную ацетоном Р, в мерную колбу. Опо-
ласкивают стакан и фильтр 30 мл ацетона Р и доводят объем фильтрата до 100,0 мл этим же растворителем. Оставляют на 24 ч без перемешивания. Используют прозрачную надосадочную жидкость в качестве испытуемого раствора.
Стандартный раствор готовят следующим образом: 0,100 г кислоты гликолевой Р, предва-
рительно высушенной под вакуумом над фосфора (V) оксидом Р, растворяют в воде Р в мерной колбе вместимостью 100 мл и доводят объем до 100,0 мл этим же растворителем. Используют раствор в течение 30 дней. Переносят 1,0 мл, 2,0 мл, 3,0 мл и 4,0 мл полученного раствора в отдельныемерныеколбы; разводятсодержимое каждой колбы до 5,0 мл водой Р, прибавляют
5 мл кислоты уксусной ледяной Р, доводят объ-
ем до 100,0 мл ацетоном Р и перемешивают. Помещают отдельно 2,0 мл испытуемого
раствора, по 2,0 мл каждого стандартного раствора и, для приготовления раствора сравнения, 2,0 мл раствора, содержащего 5 % (об/об) кис-
лоты уксусной ледяной Р и 5 % (об/об) воды Р в ацетоне Р, в отдельные мерные колбы вместимостью по 25 мл. Нагревают незакрытые колбы в течение 20 мин на водяной бане до удаления ацетона. Дают остыть и прибавляют по 5,0 мл
раствора2,7-дигидроксинафталинаРвкаждую колбу. Перемешивают, прибавляют по 15,0 мл
раствора 2,7-дигидроксинафталина Р и снова перемешивают. Закрывают колбы алюминиевой фольгой и нагревают на водяной бане в течение 20 мин. Охлаждают и доводят объем до 25,0 мл
кислотой серной Р.
Измеряют оптическую плотность (2.2.25) каждого раствора при длине волны 540 нм относительно раствора сравнения. Строят градуировочный график, используя оптические плотности стандартных растворов. По градуировочному графику и оптической плотности испытуемого раствора определяют массу (а, мг) кислоты гликолевой в испытуемом образце и рассчитывают содержание натрия гликолята по формуле:
10×1,29×a ,
(100-b)×m
где:
1,29 — коэффициент пересчета кислоты гликолевой в натрия гликолят;
b — потеря в массе при высушивании, в процентах;
m — масса испытуемого образца, в граммах.
Растворимые в воде вещества. Не более
10,0 %. 10,00 г испытуемого образца диспергируют в 800,0 мл воды Р и перемешивают по 1 мин через каждые 10 мин в течение первых 30 мин. Оставляют на 1 час и при необходимости центрифугируют.Сливаютсосадка200млнадосадочной жидкости на быстрофильтрующую фильтровальную бумагу в вакуумную фильтрационную воронку,подаютвакуумисобирают150,0млфильтрата.